

Multiple whole-genome alignments without a reference organism
- PMID: 19176791
- PMCID: PMC2665786
- DOI: 10.1101/gr.081778.108
Multiple whole-genome alignments without a reference organism
Abstract
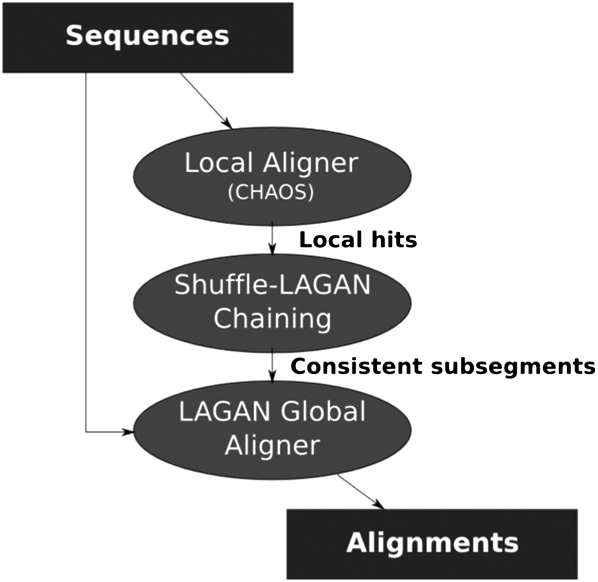
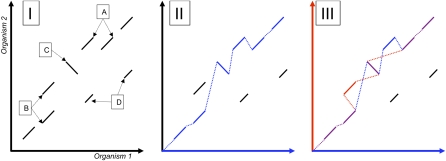
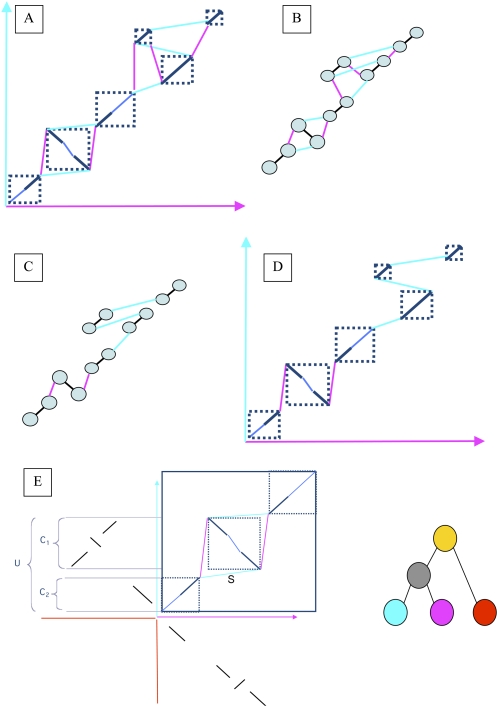
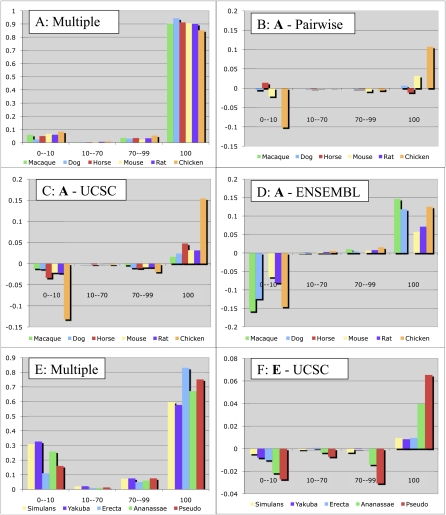
Multiple sequence alignments have become one of the most commonly used resources in genomics research. Most algorithms for multiple alignment of whole genomes rely either on a reference genome, against which all of the other sequences are laid out, or require a one-to-one mapping between the nucleotides of the genomes, preventing the alignment of recently duplicated regions. Both approaches have drawbacks for whole-genome comparisons. In this paper we present a novel symmetric alignment algorithm. The resulting alignments not only represent all of the genomes equally well, but also include all relevant duplications that occurred since the divergence from the last common ancestor. Our algorithm, implemented as a part of the VISTA Genome Pipeline (VGP), was used to align seven vertebrate and six Drosophila genomes. The resulting whole-genome alignments demonstrate a higher sensitivity and specificity than the pairwise alignments previously available through the VGP and have higher exon alignment accuracy than comparable public whole-genome alignments. Of the multiple alignment methods tested, ours performed the best at aligning genes from multigene families-perhaps the most challenging test for whole-genome alignments. Our whole-genome multiple alignments are available through the VISTA Browser at http://genome.lbl.gov/vista/index.shtml.
Figures
References
-
- Bejerano G., Pheasant M., Makunin I., Stephen S., Kent W.J., Mattick J.S., Haussler D. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials