

RosettaHoles: rapid assessment of protein core packing for structure prediction, refinement, design, and validation
- PMID: 19177366
- PMCID: PMC2708028
- DOI: 10.1002/pro.8
RosettaHoles: rapid assessment of protein core packing for structure prediction, refinement, design, and validation
Abstract
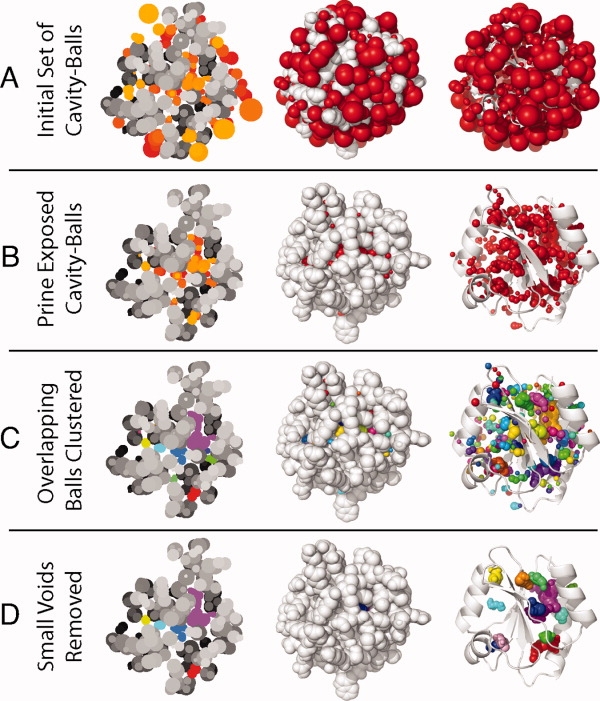
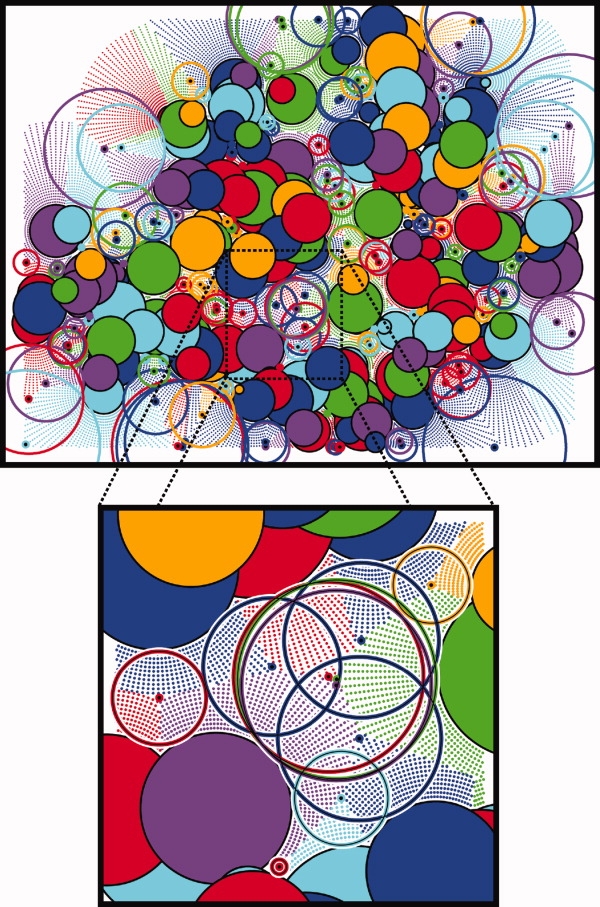
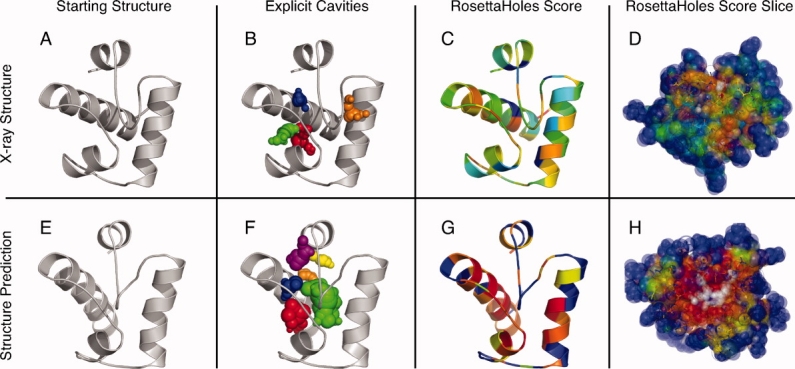
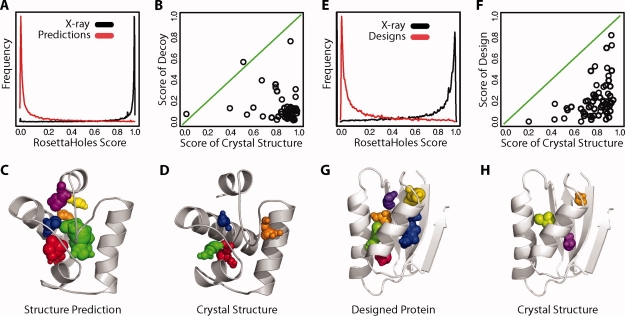
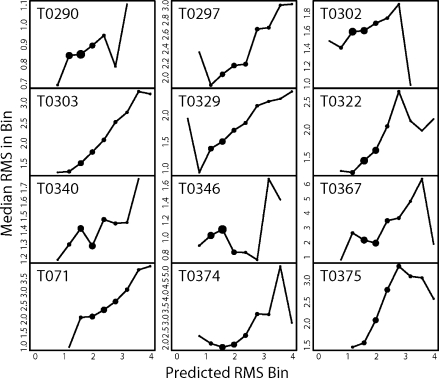
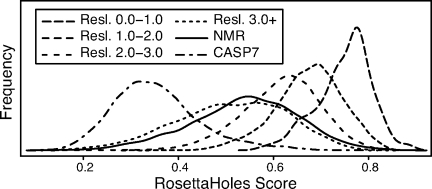
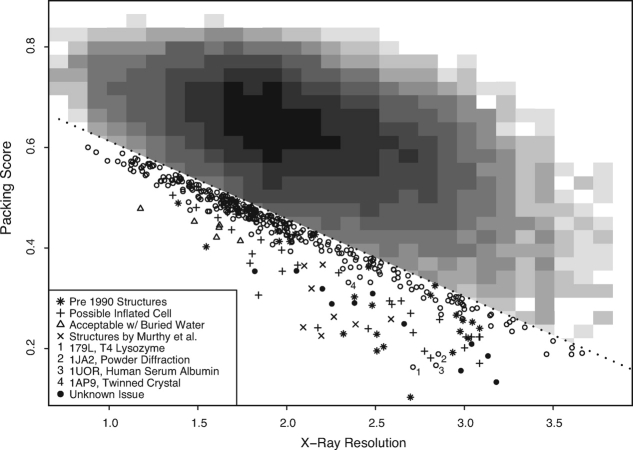
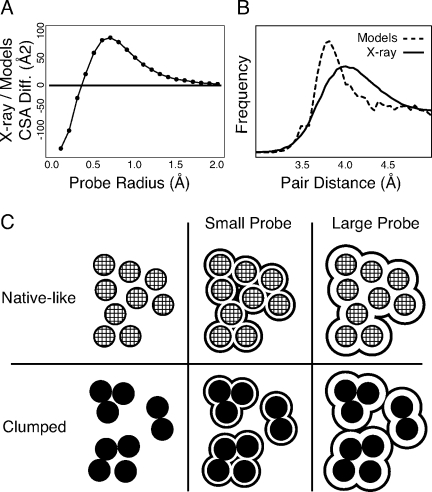
We present a novel method called RosettaHoles for visual and quantitative assessment of underpacking in the protein core. RosettaHoles generates a set of spherical cavity balls that fill the empty volume between atoms in the protein interior. For visualization, the cavity balls are aggregated into contiguous overlapping clusters and small cavities are discarded, leaving an uncluttered representation of the unfilled regions of space in a structure. For quantitative analysis, the cavity ball data are used to estimate the probability of observing a given cavity in a high-resolution crystal structure. RosettaHoles provides excellent discrimination between real and computationally generated structures, is predictive of incorrect regions in models, identifies problematic structures in the Protein Data Bank, and promises to be a useful validation tool for newly solved experimental structures.
Figures
References
-
- Kellis JTJ, Nyberg K, Fersht AR. Energetics of complementary side-chain packingin a protein hydrophobic core. Biochemistry. 1989;28:4914–4922. - PubMed
-
- Erikson AE, Baase WA, Zhang XJ, Heinz DW, Baldwin EP, Mathews BM. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 1992;255:178–183. - PubMed
-
- Eriksson AE, Baase WA, Wozniak JA, Mathews BM. A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature. 1992;355:371–373. - PubMed
-
- Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. - PubMed
-
- Liang J, Edelsbrunner H, Fu P, Sudhakar PV, Subramaniam S. Analytical shapecomputation of macromolecules: I. molecular area and volume through alpha shape. Proteins. 1998;33:1–17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
