

Discovery of amyloid-beta aggregation inhibitors using an engineered assay for intracellular protein folding and solubility
- PMID: 19177561
- PMCID: PMC2708054
- DOI: 10.1002/pro.33
Discovery of amyloid-beta aggregation inhibitors using an engineered assay for intracellular protein folding and solubility
Abstract
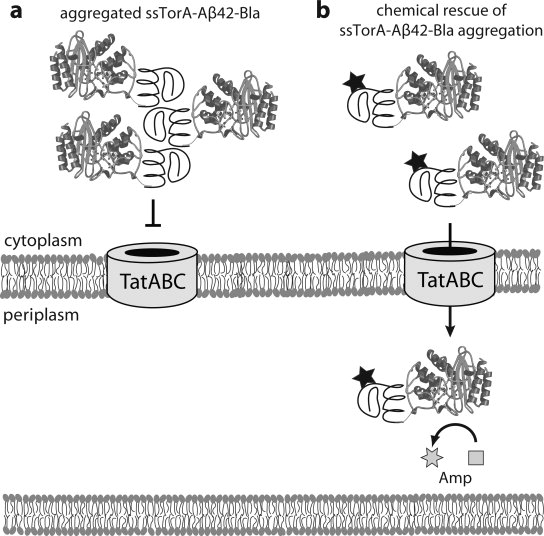
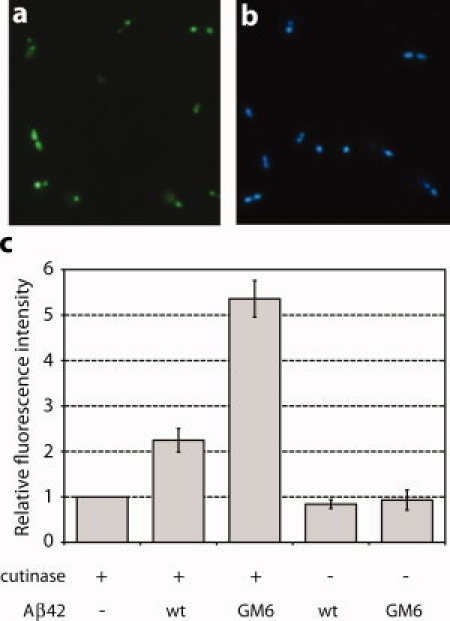
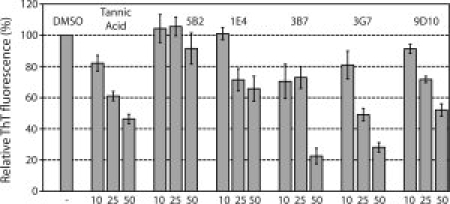
Genetic and biochemical studies suggest that Alzheimer's disease (AD) is caused by a series of events initiated by the production and subsequent aggregation of the Alzheimer's amyloid beta peptide (Abeta), the so-called amyloid cascade hypothesis. Thus, a logical approach to treating AD is the development of small molecule inhibitors that either block the proteases that generate Abeta from its precursor (beta- and gamma-secretases) or interrupt and/or reverse Abeta aggregation. To identify potent inhibitors of Abeta aggregation, we have developed a high-throughput screen based on an earlier selection that effectively paired the folding quality control feature of the Escherichia coli Tat protein export system with aggregation of the 42-residue AD pathogenesis effecter Abeta42. Specifically, a tripartite fusion between the Tat-dependent export signal ssTorA, the Abeta42 peptide and the beta-lactamase (Bla) reporter enzyme was found to be export incompetent due to aggregation of the Abeta42 moiety. Here, we reasoned that small, cell-permeable molecules that inhibited Abeta42 aggregation would render the ssTorA-Abeta42-Bla chimera competent for Tat export to the periplasm where Bla is active against beta-lactam antibiotics such as ampicillin. Using a fluorescence-based version of our assay, we screened a library of triazine derivatives and isolated four nontoxic, cell-permeable compounds that promoted efficient Tat-dependent export of ssTorA-Abeta42-Bla. Each of these was subsequently shown to be a bona fide inhibitor of Abeta42 aggregation using a standard thioflavin T fibrillization assay, thereby highlighting the utility of our bacterial assay as a useful screen for antiaggregation factors under physiological conditions.
Figures


Similar articles
-
Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway.Protein Sci. 2006 Mar;15(3):449-58. doi: 10.1110/ps.051902606. Epub 2006 Feb 1. Protein Sci. 2006. PMID: 16452624 Free PMC article.
-
A high-throughput screen for compounds that inhibit aggregation of the Alzheimer's peptide.ACS Chem Biol. 2006 Aug 22;1(7):461-9. doi: 10.1021/cb600135w. ACS Chem Biol. 2006. PMID: 17168524
-
Generic hydrophobic residues are sufficient to promote aggregation of the Alzheimer's Abeta42 peptide.Proc Natl Acad Sci U S A. 2006 Oct 24;103(43):15824-9. doi: 10.1073/pnas.0605629103. Epub 2006 Oct 12. Proc Natl Acad Sci U S A. 2006. PMID: 17038501 Free PMC article.
-
Dual inhibitors of β-amyloid aggregation and acetylcholinesterase as multi-target anti-Alzheimer drug candidates.Curr Top Med Chem. 2013;13(15):1820-42. doi: 10.2174/15680266113139990139. Curr Top Med Chem. 2013. PMID: 23931440 Review.
-
Genetic selection of solubility-enhanced proteins using the twin-arginine translocation system.Methods Mol Biol. 2011;705:53-67. doi: 10.1007/978-1-61737-967-3_4. Methods Mol Biol. 2011. PMID: 21125380 Review.
Cited by
-
Using bacterial inclusion bodies to screen for amyloid aggregation inhibitors.Microb Cell Fact. 2012 May 3;11:55. doi: 10.1186/1475-2859-11-55. Microb Cell Fact. 2012. PMID: 22553999 Free PMC article.
-
Therapeutic Potential of Direct Clearance of the Amyloid-β in Alzheimer's Disease.Brain Sci. 2020 Feb 10;10(2):93. doi: 10.3390/brainsci10020093. Brain Sci. 2020. PMID: 32050618 Free PMC article.
-
Screening strategies to identify HSP70 modulators to treat Alzheimer's disease.Drug Des Devel Ther. 2015 Jan 7;9:321-31. doi: 10.2147/DDDT.S72165. eCollection 2015. Drug Des Devel Ther. 2015. PMID: 25609918 Free PMC article. Review.
-
Mining mammalian genomes for folding competent proteins using Tat-dependent genetic selection in Escherichia coli.Protein Sci. 2009 Dec;18(12):2537-49. doi: 10.1002/pro.262. Protein Sci. 2009. PMID: 19830686 Free PMC article.
-
A genetically encoded selection for amyloid-β oligomer binders.Nat Chem Biol. 2025 Jul 15. doi: 10.1038/s41589-025-01975-4. Online ahead of print. Nat Chem Biol. 2025. PMID: 40664790
References
-
- Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. - PubMed
-
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. - PubMed
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. - PubMed
-
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. - PMC - PubMed
-
- Aisen PS. The development of anti-amyloid therapy for Alzheimer's disease: from secretase modulators to polymerisation inhibitors. CNS Drugs. 2005;19:989–996. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
