

The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload
- PMID: 19179290
- PMCID: PMC2650158
- DOI: 10.1073/pnas.0813013106
The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload
Abstract
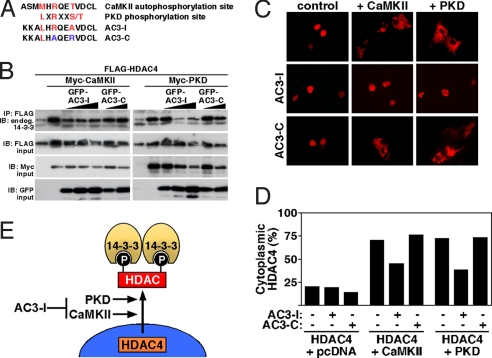
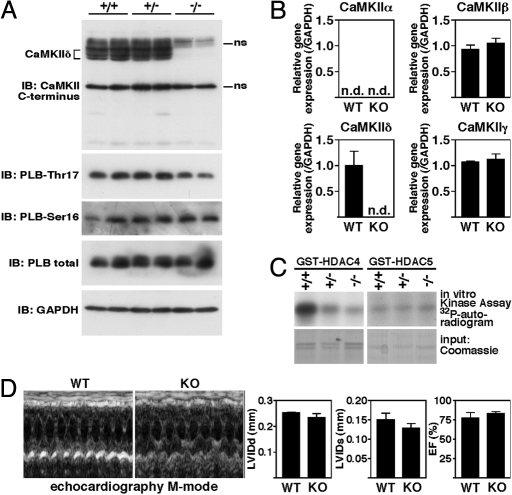
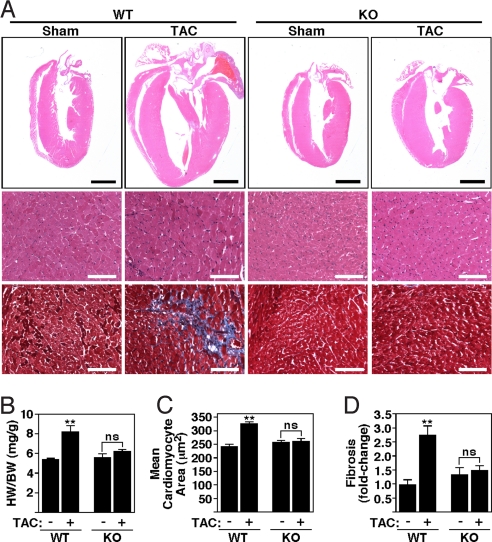
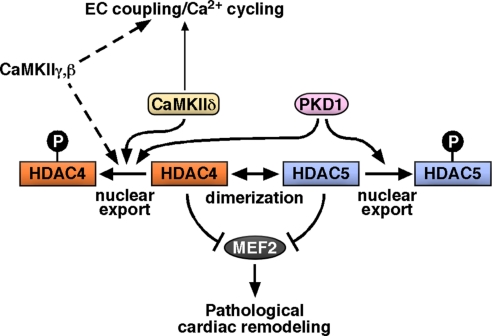
Acute and chronic injuries to the heart result in perturbation of intracellular calcium signaling, which leads to pathological cardiac hypertrophy and remodeling. Calcium/calmodulin-dependent protein kinase II (CaMKII) has been implicated in the transduction of calcium signals in the heart, but the specific isoforms of CaMKII that mediate pathological cardiac signaling have not been fully defined. To investigate the potential involvement in heart disease of CaMKIIdelta, the major CaMKII isoform expressed in the heart, we generated CaMKIIdelta-null mice. These mice are viable and display no overt abnormalities in cardiac structure or function in the absence of stress. However, pathological cardiac hypertrophy and remodeling are attenuated in response to pressure overload in these animals. Cardiac extracts from CaMKIIdelta-null mice showed diminished kinase activity toward histone deacetylase 4 (HDAC4), a substrate of stress-responsive protein kinases and suppressor of stress-dependent cardiac remodeling. In contrast, phosphorylation of the closely related HDAC5 was unaffected in hearts of CaMKIIdelta-null mice, underscoring the specificity of the CaMKIIdelta signaling pathway for HDAC4 phosphorylation. We conclude that CaMKIIdelta functions as an important transducer of stress stimuli involved in pathological cardiac remodeling in vivo, which is mediated, at least in part, by the phosphorylation of HDAC4. These findings point to CaMKIIdelta as a potential therapeutic target for the maintenance of cardiac function in the setting of pressure overload.
Conflict of interest statement
Conflict of interest statement: E.N.O. is cofounder of MiRagen Therapeutics. E.N.O. and J.B. are consultants for Gilead Therapeutics and have filed a patent on the modulation of cardiac hypertrophy and CaMKII.
Figures



References
-
- Frey N, Olson EN. Cardiac hypertrophy: The good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. - PubMed
-
- Frey N, McKinsey TA, Olson EN. Decoding calcium signals involved in cardiac growth and function. Nat Med. 2000;6:1221–1227. - PubMed
-
- McKinsey TA. Derepression of pathological cardiac genes by members of the CaM kinase superfamily. Cardiovasc Res. 2007;73:667–677. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
