

Recombineering: a homologous recombination-based method of genetic engineering
- PMID: 19180090
- PMCID: PMC2790811
- DOI: 10.1038/nprot.2008.227
Recombineering: a homologous recombination-based method of genetic engineering
Abstract
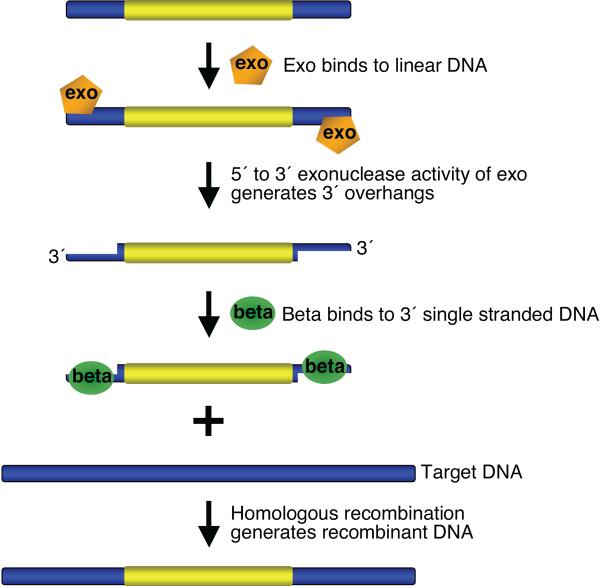
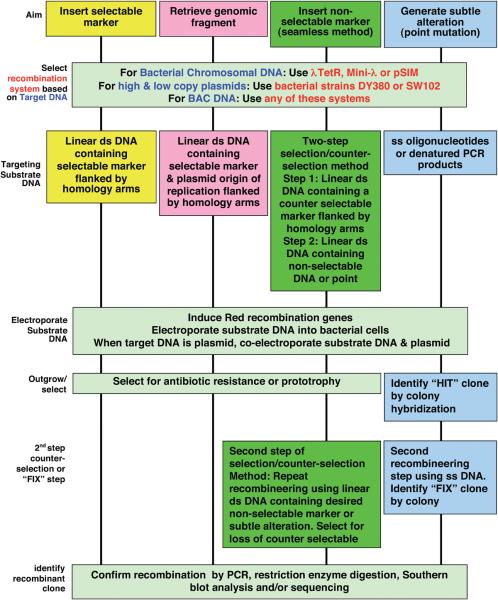
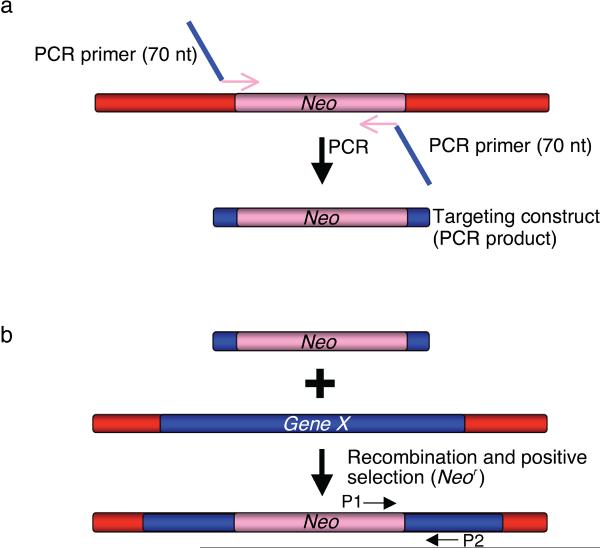
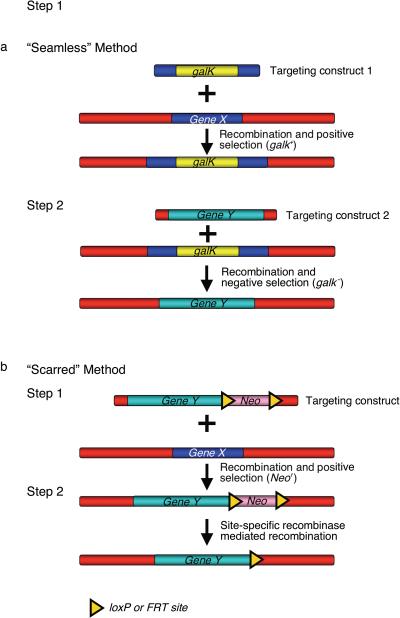
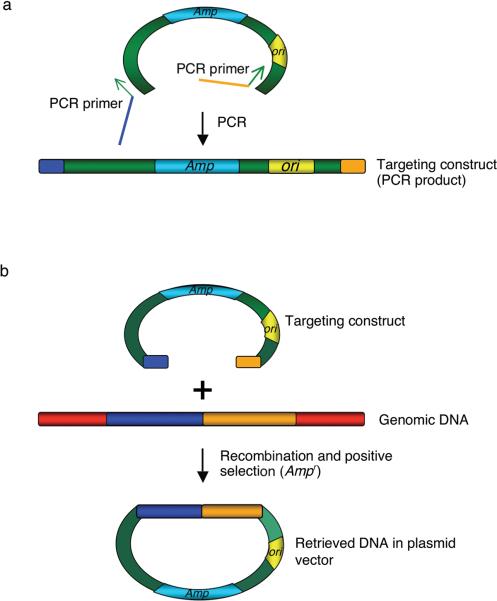
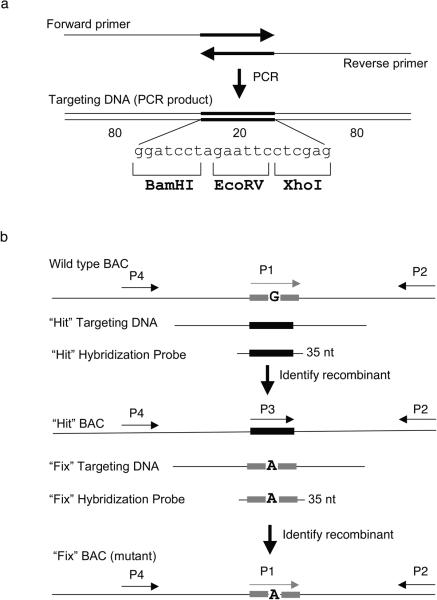
Recombineering is an efficient method of in vivo genetic engineering applicable to chromosomal as well as episomal replicons in Escherichia coli. This method circumvents the need for most standard in vitro cloning techniques. Recombineering allows construction of DNA molecules with precise junctions without constraints being imposed by restriction enzyme site location. Bacteriophage homologous recombination proteins catalyze these recombineering reactions using double- and single-stranded linear DNA substrates, so-called targeting constructs, introduced by electroporation. Gene knockouts, deletions and point mutations are readily made, gene tags can be inserted and regions of bacterial artificial chromosomes or the E. coli genome can be subcloned by gene retrieval using recombineering. Most of these constructs can be made within about 1 week's time.
Figures

References
-
- Copeland NG, Jenkins NA, Court DL. Recombineering: a powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2001;2:769–779. - PubMed
-
- Zhang Y, Buchholz F, Muyrers JP, Stewart AF. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 1998;20:123–128. - PubMed
-
- Zhang Y, Muyrers JP, Testa G, Stewart AF. DNA cloning by homologous recombination in Escherichia coli. Nat. Biotechnol. 2000;18:1314–1317. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
