

beta2-glycoprotein i is a cofactor for tissue plasminogen activator-mediated plasminogen activation
- PMID: 19180513
- PMCID: PMC2965030
- DOI: 10.1002/art.24262
beta2-glycoprotein i is a cofactor for tissue plasminogen activator-mediated plasminogen activation
Abstract
Objective: Regulation of the conversion of plasminogen to plasmin by tissue plasminogen activator (tPA) is critical in the control of fibrin deposition. While several plasminogen activators have been described, soluble plasma cofactors that stimulate fibrinolysis have not been characterized. The purpose of this study was to investigate the effects of beta(2)-glycoprotein I (beta(2)GPI), an abundant plasma glycoprotein, on tPA-mediated plasminogen activation.
Methods: The effect of beta(2)GPI on tPA-mediated activation of plasminogen was assessed using amidolytic assays, a fibrin gel, and plasma clots. Binding of beta(2)GPI to tPA and plasminogen was determined in parallel. The effects of IgG fractions and anti-beta(2)GPI antibodies from patients with antiphospholipid syndrome (APS) on tPA-mediated plasminogen activation were also measured.
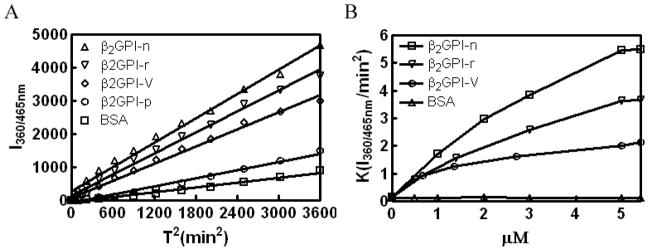
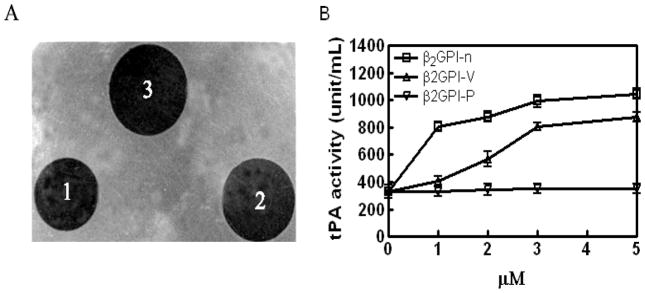
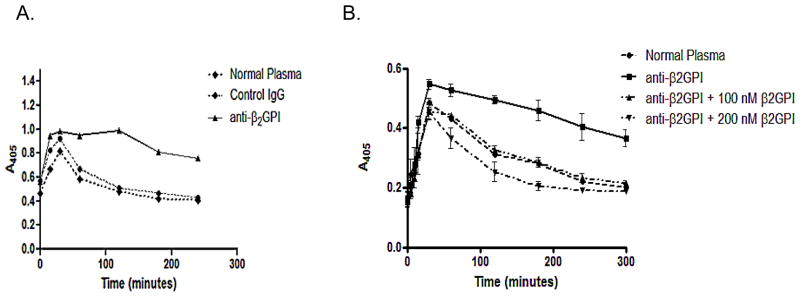
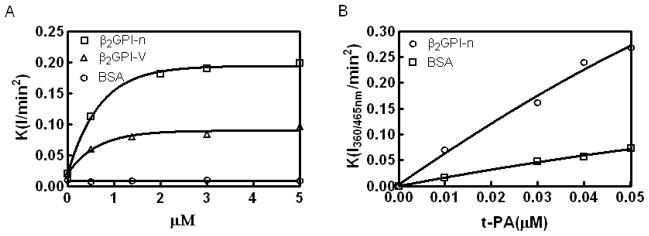
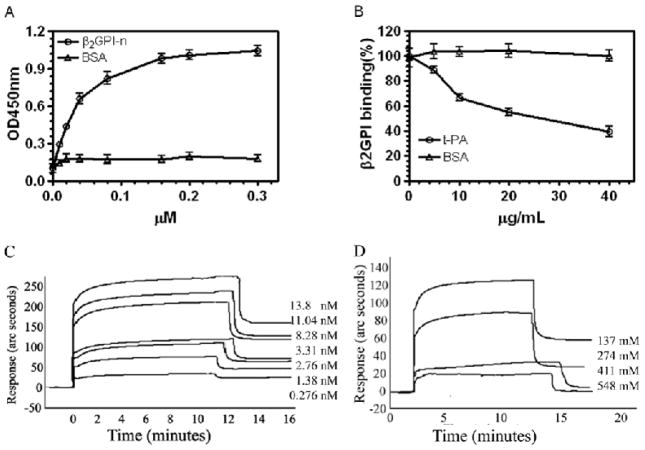
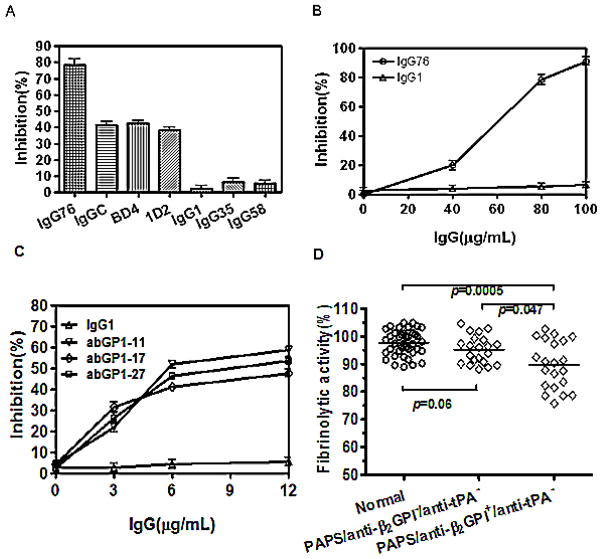
Results: Beta(2)-glycoprotein I stimulated tPA-dependent plasminogen activation in the fluid phase and within a fibrin gel. The beta(2)GPI region responsible for stimulating tPA activity was shown to be at least partly contained within beta(2)GPI domain V. In addition, beta(2)GPI bound tPA with high affinity (K(d) approximately 20 nM), stimulated tPA amidolytic activity, and caused an overall 20-fold increase in the catalytic efficiency (K(cat)/K(m)) of tPA-mediated conversion of Glu-plasminogen to plasmin. Moreover, depletion of beta(2)GPI from plasma led to diminished rates of clot lysis, with restoration of normal lysis rates following beta(2)GPI repletion. Stimulation of tPA-mediated plasminogen activity by beta(2)GPI was inhibited by monoclonal anti-beta(2)GPI antibodies as well as by anti-beta(2)GPI antibodies from patients with APS.
Conclusion: These findings suggest that beta(2)GPI may be an endogenous regulator of fibrinolysis. Impairment of beta(2)GPI-stimulated fibrinolysis by anti-beta(2)GPI antibodies may contribute to the development of thrombosis in patients with APS.
Figures
Similar articles
-
Beta2-glycoprotein I, anti-beta2-glycoprotein I, and fibrinolysis.Thromb Res. 2004;114(5-6):461-5. doi: 10.1016/j.thromres.2004.07.013. Thromb Res. 2004. PMID: 15507279 Review.
-
Effects of beta2-glycoprotein I and monoclonal anticardiolipin antibodies on extrinsic fibrinolysis.Semin Thromb Hemost. 2000;26(1):85-90. doi: 10.1055/s-2000-9808. Semin Thromb Hemost. 2000. PMID: 10805287
-
Impaired fibrinolysis in the antiphospholipid syndrome.Curr Rheumatol Rep. 2010 Feb;12(1):53-7. doi: 10.1007/s11926-009-0075-4. Curr Rheumatol Rep. 2010. PMID: 20425534 Free PMC article. Review.
-
Pathogenesis of antiphospholipid antibodies: impairment of fibrinolysis and monocyte activation via the p38 mitogen-activated protein kinase pathway.Immunobiology. 2005;210(10):775-80. doi: 10.1016/j.imbio.2005.10.009. Epub 2005 Oct 21. Immunobiology. 2005. PMID: 16325497 Review.
-
Modulation of fibrinolysis by the combined action of phospholipids and immunoglobulins.Blood Coagul Fibrinolysis. 2008 Jan;19(1):82-8. doi: 10.1097/MBC.0b013e3282f38c6f. Blood Coagul Fibrinolysis. 2008. PMID: 18180621
Cited by
-
Determining Thrombogenicity: Using a Modified Thrombin Generation Assay to Detect the Level of Thrombotic Event Risk in Lupus Anticoagulant-Positive Patients.Biomedicines. 2023 Dec 16;11(12):3329. doi: 10.3390/biomedicines11123329. Biomedicines. 2023. PMID: 38137550 Free PMC article.
-
The Relationship Between Thrombo-Inflammatory Biomarkers and Cellular Indices of Inflammation in Lymphoma Patients.Clin Appl Thromb Hemost. 2021 Jan-Dec;27:10760296211050358. doi: 10.1177/10760296211050358. Clin Appl Thromb Hemost. 2021. PMID: 34713728 Free PMC article.
-
Autoimmunity and recurrent pregnancy losses.Clin Rev Allergy Immunol. 2010 Dec;39(3):148-52. doi: 10.1007/s12016-009-8179-1. Clin Rev Allergy Immunol. 2010. PMID: 19842070 Review.
-
Antiphospholipid antibodies in critically ill COVID-19 patients with thromboembolism: cause of disease or epiphenomenon?J Thromb Thrombolysis. 2021 Aug;52(2):542-552. doi: 10.1007/s11239-021-02470-y. Epub 2021 May 10. J Thromb Thrombolysis. 2021. PMID: 33973157 Free PMC article. Review.
-
Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS.Blood. 2020 Jan 23;135(4):239-251. doi: 10.1182/blood.2019003863. Blood. 2020. PMID: 31812994 Free PMC article.
References
-
- Polz E, Kostner GM. The binding of β2-glycoprotein I to human serum lipoproteins. FEBS Lett. 1979;102:183–6. - PubMed
-
- Kamboh MI, Mehdi H. Genetics of apolipoprotein H (beta 2-glycoprotein I) and anionic phospholipid binding. Lupus. 1998;7 (Suppl 2):S10–S13. - PubMed
-
- Ohkura N, Hagihara Y, Yoshimura T, Goto Y, Kato H. Plasmin can reduce the function of human beta 2 glycoprotein I by cleaving domain V into a nicked form. Blood. 1998;91:4173–9. - PubMed
-
- Galli M. Antiphospholipid syndrome: association between laboratory tests and clinical practice. Pathophysiol Haemost Thromb. 2003;33(5–6):249–55. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
