

Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1
- PMID: 19181672
- PMCID: PMC2724920
- DOI: 10.1093/brain/awn325
Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1
Abstract
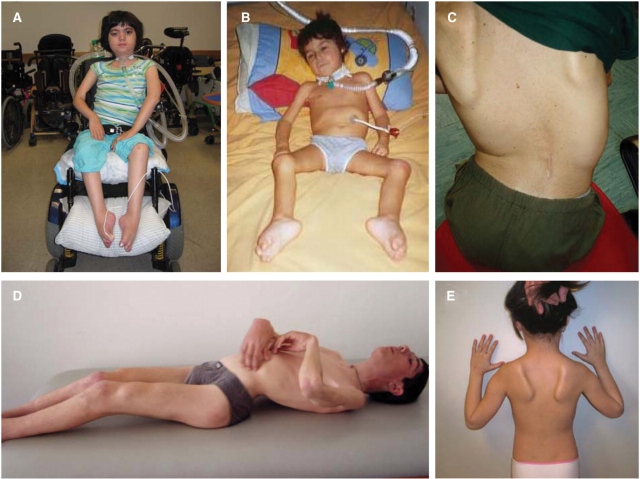
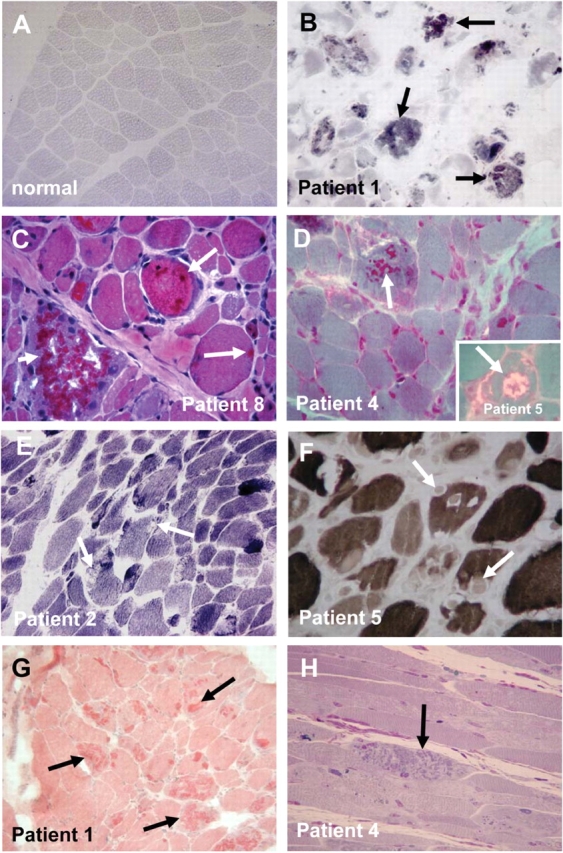
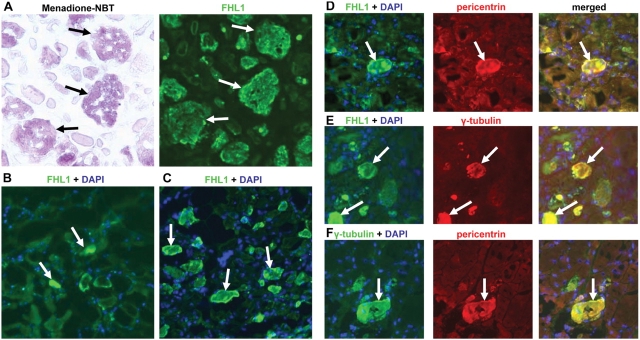
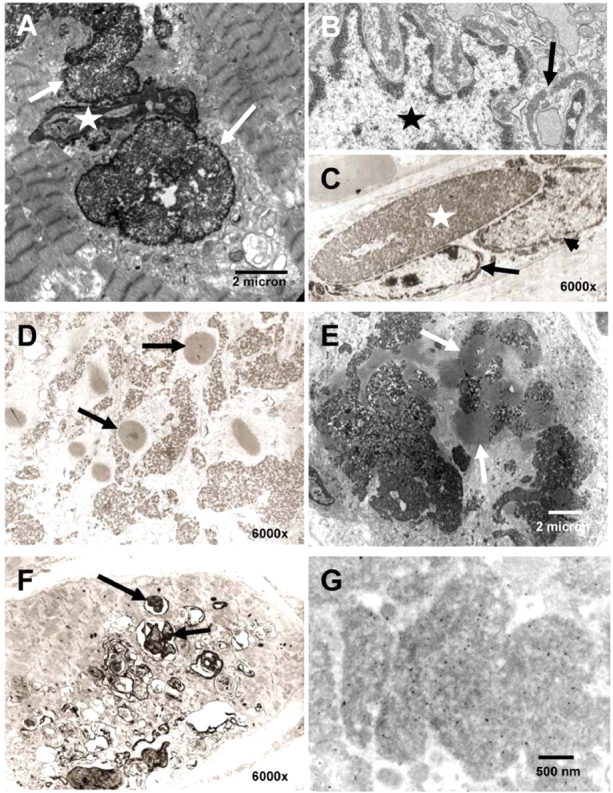
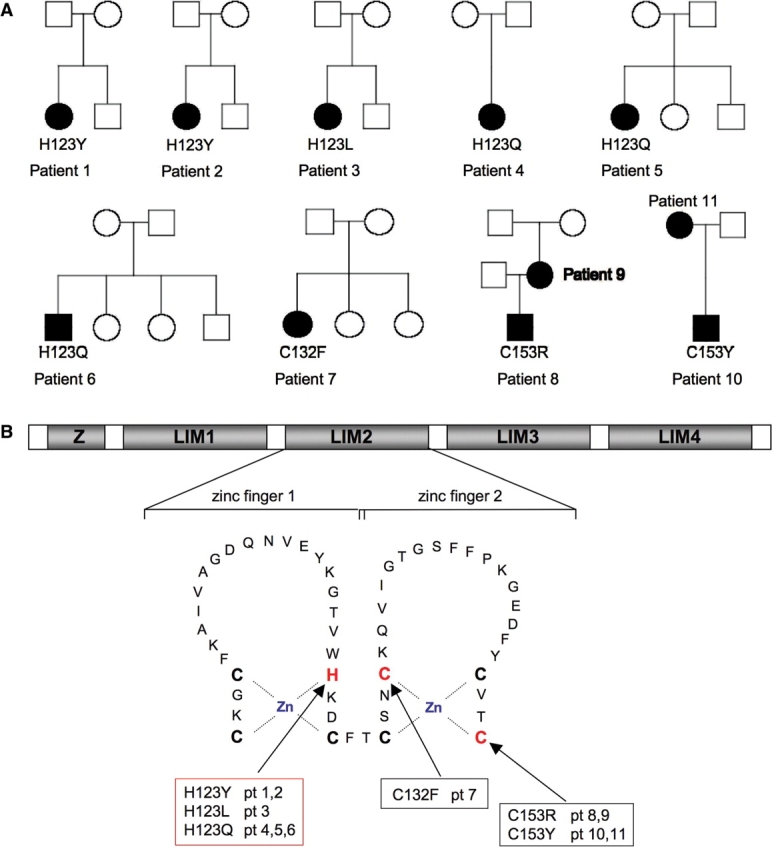
We recently identified the X-chromosomal four and a half LIM domain gene FHL1 as the causative gene for reducing body myopathy, a disorder characterized by progressive weakness and intracytoplasmic aggregates in muscle that exert reducing activity on menadione nitro-blue-tetrazolium (NBT). The mutations detected in FHL1 affected highly conserved zinc coordinating residues within the second LIM domain and lead to the formation of aggregates when transfected into cells. Our aim was to define the clinical and morphological phenotype of this myopathy and to assess the mutational spectrum of FHL1 mutations in reducing body myopathy in a larger cohort of patients. Patients were ascertained via the detection of reducing bodies in muscle biopsy sections stained with menadione-NBT followed by clinical, histological, ultrastructural and molecular genetic analysis. A total of 11 patients from nine families were included in this study, including seven sporadic patients with early childhood onset disease and four familial cases with later onset. Weakness in all patients was progressive, sometimes rapidly so. Respiratory failure was common and scoliosis and spinal rigidity were significant in some of the patients. Analysis of muscle biopsies confirmed the presence of aggregates of FHL1 positive material in all biopsies. In two patients in whom sequential biopsies were available the aggregate load in muscle sections appeared to increase over time. Ultrastructural analysis revealed that cytoplasmic bodies were regularly seen in conjunction with the reducing bodies. The mutations detected were exclusive to the second LIM domain of FHL1 and were found in both sporadic as well as familial cases of reducing body myopathy. Six of the nine mutations affected the crucial zinc coordinating residue histidine 123. All mutations in this residue were de novo and were associated with a severe clinical course, in particular in one male patient (H123Q). Mutations in the zinc coordinating residue cysteine 153 were associated with a milder phenotype and were seen in the familial cases in which the boys were still more severely affected compared to their mothers. We expect the mild end of the spectrum to significantly expand in the future. On the severe end of the spectrum we define reducing body myopathy as a progressive disease with early, but not necessarily congenital onset, distinguishing this condition from the classic essentially non-progressive congenital myopathies.
Figures
References
-
- Bertini E, Salviati G, Apollo F, Ricci E, Servidei S, Broccolini A, et al. Reducing body myopathy and desmin storage in skeletal muscle: morphological and biochemical findings. Acta Neuropathol (Berl) 1994;87:106–12. - PubMed
-
- Brooke MH, Neville HE. Reducing body myopathy. Neurology. 1972;22:829–40. - PubMed
-
- Brown S, McGrath MJ, Ooms LM, Gurung R, Maimone MM, Mitchell CA. Characterization of two isoforms of the skeletal muscle LIM protein 1, SLIM1. Localization of SLIM1 at focal adhesions and the isoform slimmer in the nucleus of myoblasts and cytoplasm of myotubes suggests distinct roles in the cytoskeleton and in nuclear-cytoplasmic communication. J Biol Chem. 1999;274:27083–91. - PubMed
-
- Carpenter S, Karpati G, Holland P. New observations in reducing body myopathy. Neurology. 1985;35:818–27. - PubMed
-
- Curtiss J, Heilig JS. DeLIMiting development. Bioessays. 1998;20:58–69. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
