

Ab initio and template-based prediction of multi-class distance maps by two-dimensional recursive neural networks
- PMID: 19183478
- PMCID: PMC2654788
- DOI: 10.1186/1472-6807-9-5
Ab initio and template-based prediction of multi-class distance maps by two-dimensional recursive neural networks
Abstract
Background: Prediction of protein structures from their sequences is still one of the open grand challenges of computational biology. Some approaches to protein structure prediction, especially ab initio ones, rely to some extent on the prediction of residue contact maps. Residue contact map predictions have been assessed at the CASP competition for several years now. Although it has been shown that exact contact maps generally yield correct three-dimensional structures, this is true only at a relatively low resolution (3-4 A from the native structure). Another known weakness of contact maps is that they are generally predicted ab initio, that is not exploiting information about potential homologues of known structure.
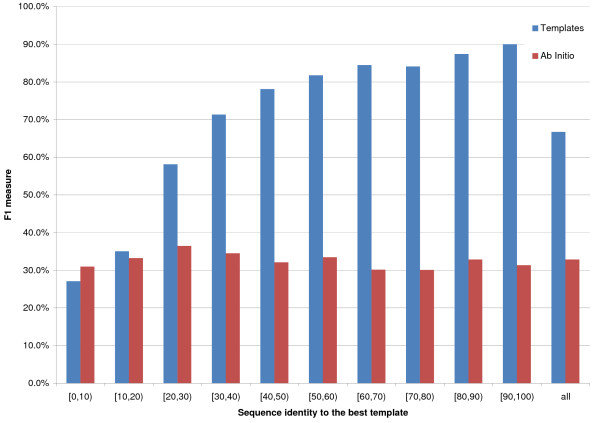
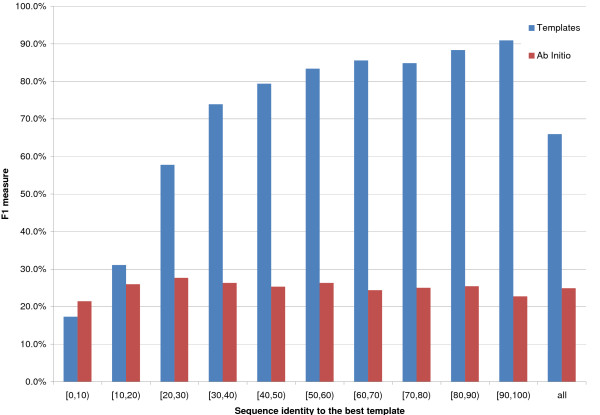
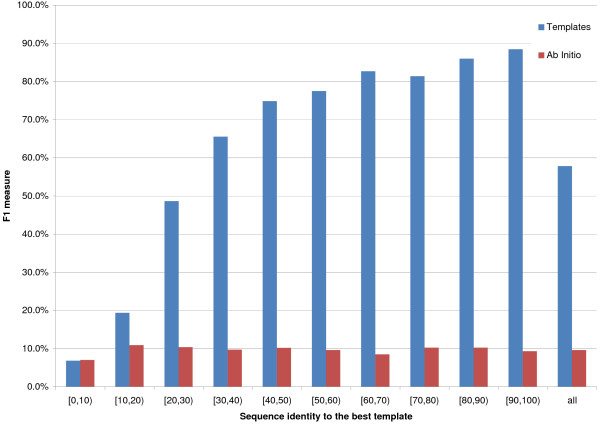
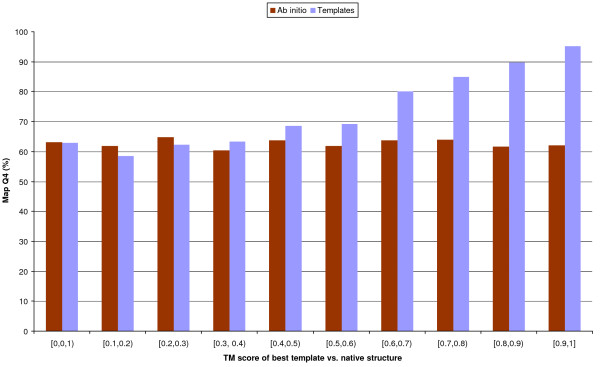
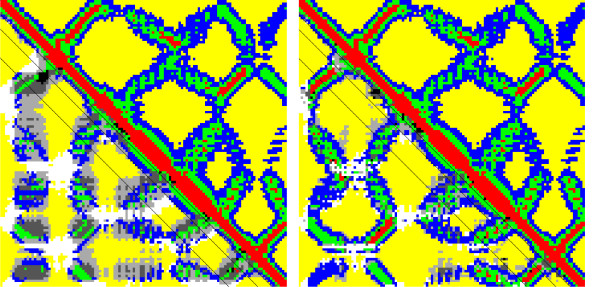
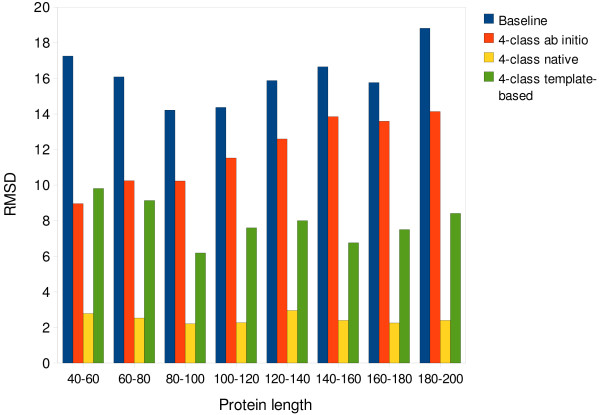
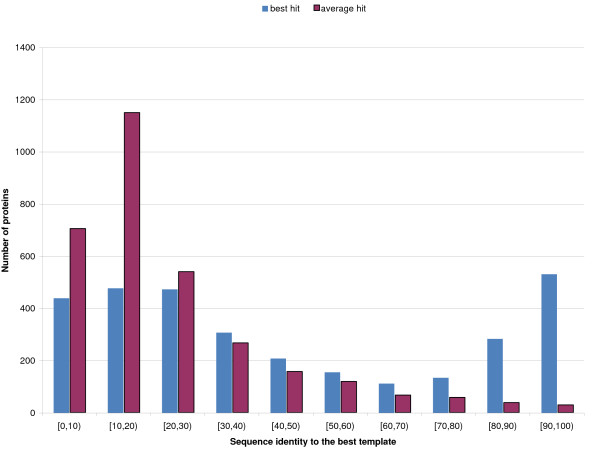
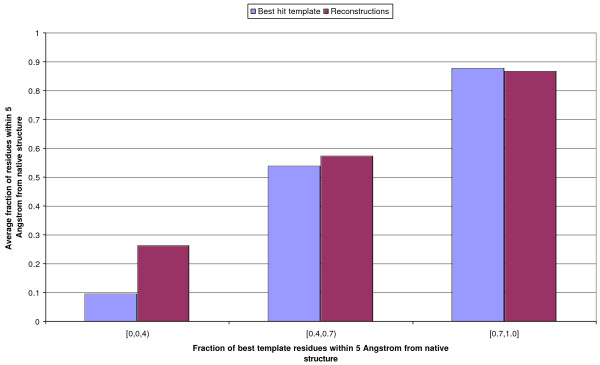
Results: We introduce a new class of distance restraints for protein structures: multi-class distance maps. We show that C alpha trace reconstructions based on 4-class native maps are significantly better than those from residue contact maps. We then build two predictors of 4-class maps based on recursive neural networks: one ab initio, or relying on the sequence and on evolutionary information; one template-based, or in which homology information to known structures is provided as a further input. We show that virtually any level of sequence similarity to structural templates (down to less than 10%) yields more accurate 4-class maps than the ab initio predictor. We show that template-based predictions by recursive neural networks are consistently better than the best template and than a number of combinations of the best available templates. We also extract binary residue contact maps at an 8 A threshold (as per CASP assessment) from the 4-class predictors and show that the template-based version is also more accurate than the best template and consistently better than the ab initio one, down to very low levels of sequence identity to structural templates. Furthermore, we test both ab-initio and template-based 8 A predictions on the CASP7 targets using a pre-CASP7 PDB, and find that both predictors are state-of-the-art, with the template-based one far outperforming the best CASP7 systems if templates with sequence identity to the query of 10% or better are available. Although this is not the main focus of this paper we also report on reconstructions of C alpha traces based on both ab initio and template-based 4-class map predictions, showing that the latter are generally more accurate even when homology is dubious.
Conclusion: Accurate predictions of multi-class maps may provide valuable constraints for improved ab initio and template-based prediction of protein structures, naturally incorporate multiple templates, and yield state-of-the-art binary maps. Predictions of protein structures and 8 A contact maps based on the multi-class distance map predictors described in this paper are freely available to academic users at the url http://distill.ucd.ie/.
Figures
Similar articles
-
Toward an accurate prediction of inter-residue distances in proteins using 2D recursive neural networks.BMC Bioinformatics. 2014 Jan 10;15:6. doi: 10.1186/1471-2105-15-6. BMC Bioinformatics. 2014. PMID: 24410833 Free PMC article.
-
Ab initio and homology based prediction of protein domains by recursive neural networks.BMC Bioinformatics. 2009 Jun 26;10:195. doi: 10.1186/1471-2105-10-195. BMC Bioinformatics. 2009. PMID: 19558651 Free PMC article.
-
A two-stage approach for improved prediction of residue contact maps.BMC Bioinformatics. 2006 Mar 30;7:180. doi: 10.1186/1471-2105-7-180. BMC Bioinformatics. 2006. PMID: 16573808 Free PMC article.
-
What method to use for protein-protein docking?Curr Opin Struct Biol. 2019 Apr;55:1-7. doi: 10.1016/j.sbi.2018.12.010. Epub 2019 Feb 1. Curr Opin Struct Biol. 2019. PMID: 30711743 Free PMC article. Review.
-
A glance into the evolution of template-free protein structure prediction methodologies.Biochimie. 2020 Aug;175:85-92. doi: 10.1016/j.biochi.2020.04.026. Epub 2020 May 15. Biochimie. 2020. PMID: 32417458 Review.
Cited by
-
OMPcontact: An Outer Membrane Protein Inter-Barrel Residue Contact Prediction Method.J Comput Biol. 2017 Mar;24(3):217-228. doi: 10.1089/cmb.2015.0236. Epub 2016 Aug 11. J Comput Biol. 2017. PMID: 27513917 Free PMC article.
-
Recent Applications of Deep Learning Methods on Evolution- and Contact-Based Protein Structure Prediction.Int J Mol Sci. 2021 Jun 2;22(11):6032. doi: 10.3390/ijms22116032. Int J Mol Sci. 2021. PMID: 34199677 Free PMC article. Review.
-
Accurate Ab Initio and Template-Based Prediction of Short Intrinsically-Disordered Regions by Bidirectional Recurrent Neural Networks Trained on Large-Scale Datasets.Int J Mol Sci. 2015 Aug 21;16(8):19868-85. doi: 10.3390/ijms160819868. Int J Mol Sci. 2015. PMID: 26307973 Free PMC article.
-
SCLpredT: Ab initio and homology-based prediction of subcellular localization by N-to-1 neural networks.Springerplus. 2013 Oct 3;2:502. doi: 10.1186/2193-1801-2-502. eCollection 2013. Springerplus. 2013. PMID: 24133649 Free PMC article.
-
Toward an accurate prediction of inter-residue distances in proteins using 2D recursive neural networks.BMC Bioinformatics. 2014 Jan 10;15:6. doi: 10.1186/1471-2105-15-6. BMC Bioinformatics. 2014. PMID: 24410833 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
