

What determines the structure and stability of KFFE monomers, dimers, and protofibrils?
- PMID: 19186127
- PMCID: PMC2716633
- DOI: 10.1016/j.bpj.2008.10.040
What determines the structure and stability of KFFE monomers, dimers, and protofibrils?
Abstract
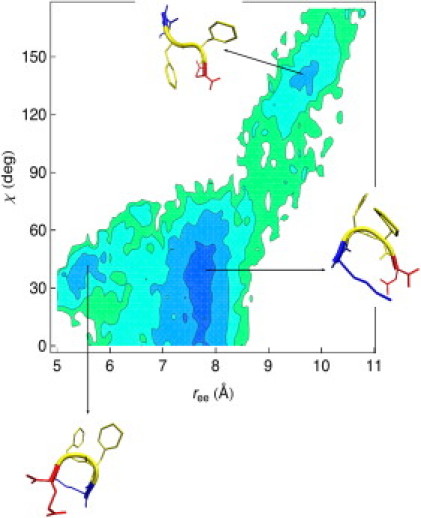
The self-assembly of the KFFE peptide was studied using replica exchange molecular dynamics simulations with a fully atomic description of the peptide and explicit solvent. The relative roles of the aromatic residues and oppositely charged end groups in stabilizing the earliest oligomers and the end-products of aggregation were investigated. beta and non-beta-peptide conformations compete in the monomeric state as a result of a balancing between the high beta-sheet propensity of the phenylalanine residues and charge-charge interactions that favor non-beta-conformations. Dimers are present in beta- and non-beta-sheet conformations and are stabilized primarily by direct and water-mediated charge-charge interactions between oppositely charged side chains and between oppositely charged termini, with forces between aromatic residues playing a minor role. Dimerization to a beta-sheet, fibril-competent state, is seen to be a cooperative process, with the association process inducing beta-structure in otherwise non-beta-monomers. We propose a model for the KFFE fibril, with mixed interface and antiparallel sheet and strand arrangements, which is consistent with experimental electron microscopy measurements. Both aromatic and charge-charge interactions contribute to the fibril stability, although the dominant contribution arises from electrostatic interactions.
Figures






References
-
- Dobson C.M. Protein folding and misfolding. Nature. 2003;426:884–890. - PubMed
-
- Makin O.S., Serpell L.C. X-ray diffraction studies of amyloid structure. Methods Mol. Biol. 2005;299:67–80. - PubMed
-
- Meredith S. Protein denaturation and aggregation—cellular responses to denatured and aggregated proteins. Cell Injury: Mech. Resp. Repair. 2005;1066:181–221. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
