

Secondary consequences of beta cell inexcitability: identification and prevention in a murine model of K(ATP)-induced neonatal diabetes mellitus
- PMID: 19187772
- PMCID: PMC4793729
- DOI: 10.1016/j.cmet.2008.12.005
Secondary consequences of beta cell inexcitability: identification and prevention in a murine model of K(ATP)-induced neonatal diabetes mellitus
Abstract
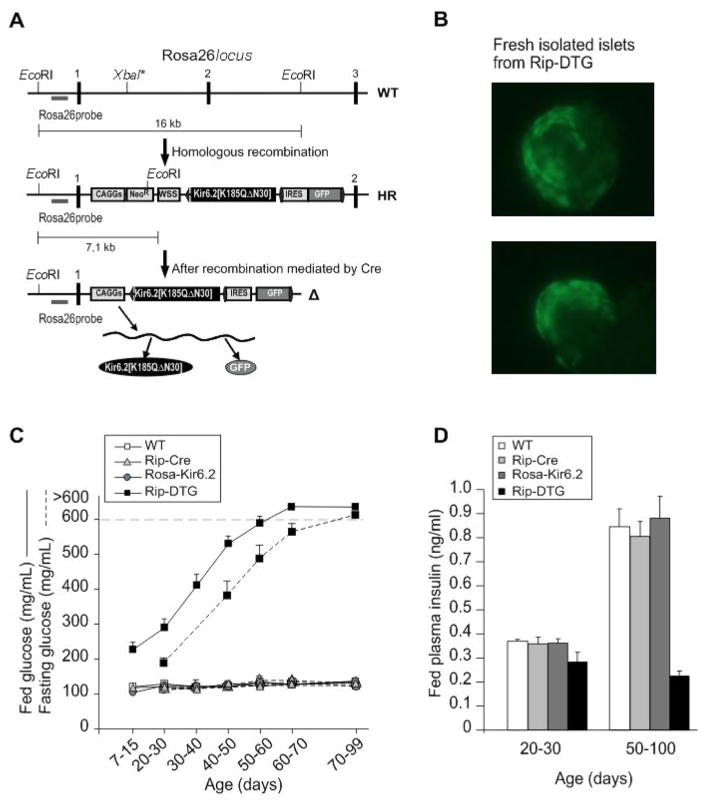
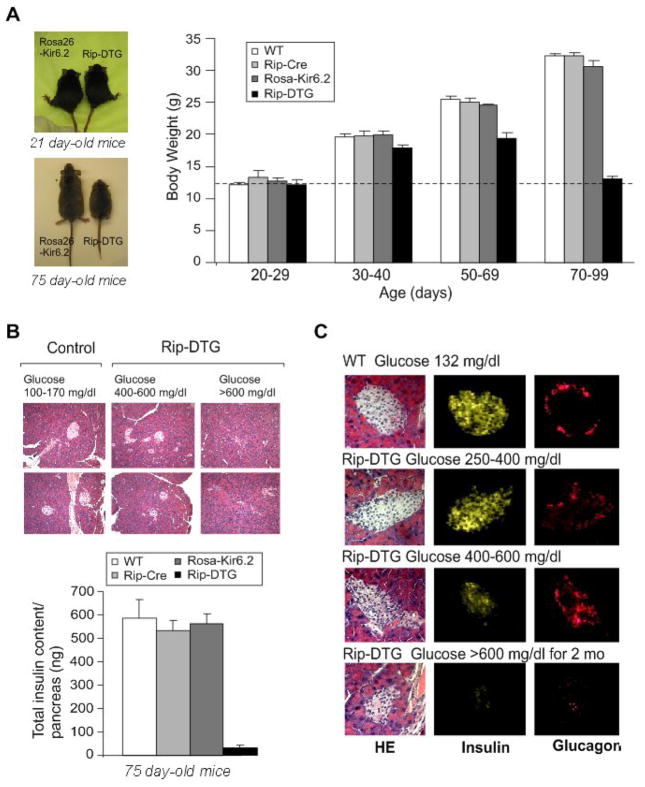
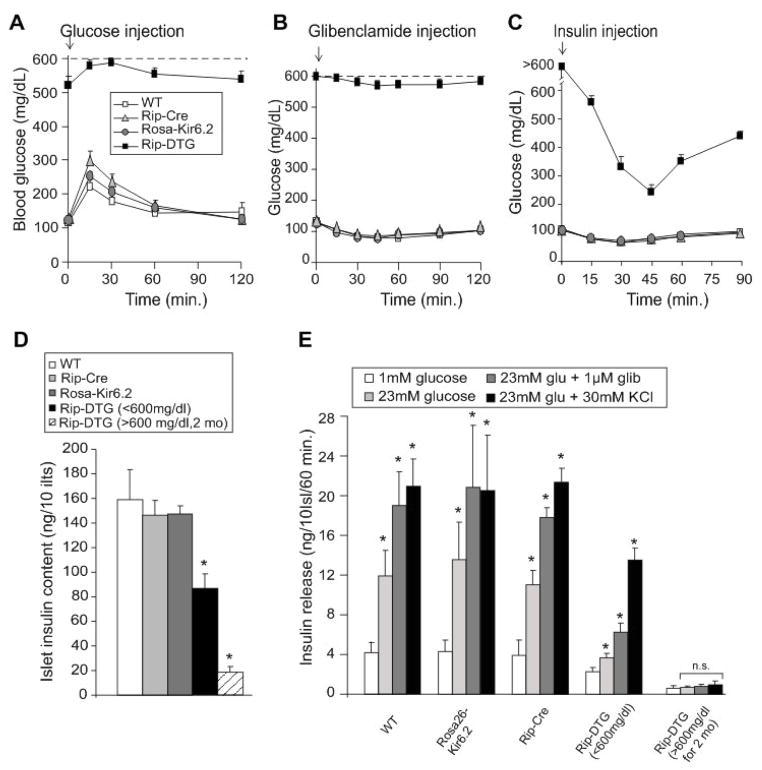
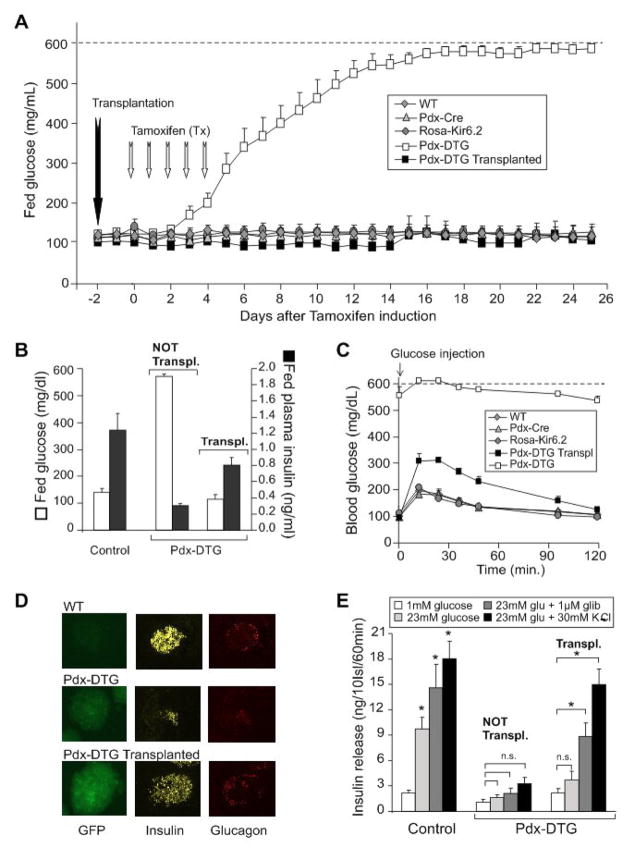
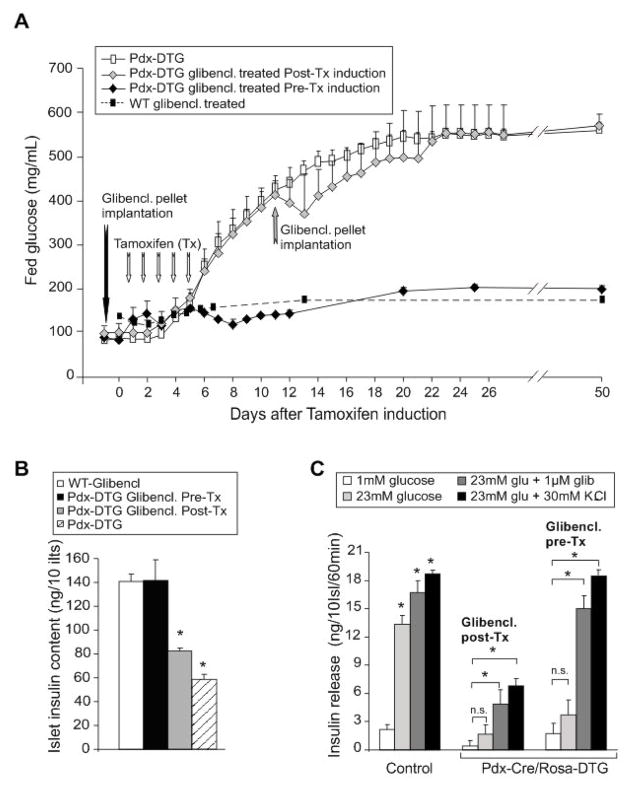
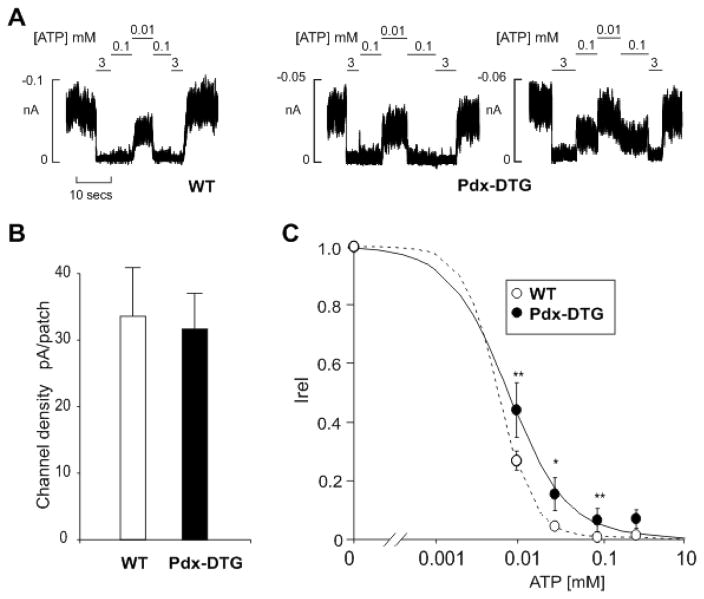
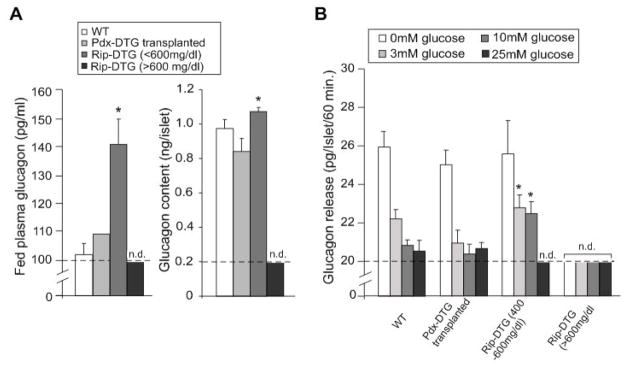
ATP-insensitive K(ATP) channel mutations cause neonatal diabetes mellitus (NDM). To explore the mechanistic etiology, we generated transgenic mice carrying an ATP-insensitive mutant K(ATP) channel subunit. Constitutive expression in pancreatic beta cells caused neonatal hyperglycemia and progression to severe diabetes and growth retardation, with loss of islet insulin content and beta cell architecture. Tamoxifen-induced expression in adult beta cells led to diabetes within 2 weeks, with similar secondary consequences. Diabetes was prevented by transplantation of normal islets under the kidney capsule. Moreover, the endogenous islets maintained normal insulin content and secretion in response to sulfonylureas, but not glucose, consistent with reduced ATP sensitivity of beta cell K(ATP) channels. In NDM, transfer to sulfonylurea therapy is less effective in older patients. This may stem from poor glycemic control or lack of insulin because glibenclamide treatment prior to tamoxifen induction prevented diabetes and secondary complications in mice but failed to halt disease progression after diabetes had developed.
Figures
References
-
- Ashcroft FM, Gribble FM. ATP-sensitive K+ channels and insulin secretion: their role in health and disease.[comment] Diabetologia. 1999;42:903–919. - PubMed
-
- Bansal P, Wang Q. Insulin as a physiological modulator of glucagon secretion. Am J Physiol Endocrinol Metab. 2008;295:E751–761. - PubMed
-
- Del Prato S, Marchetti P. Beta- and alpha-cell dysfunction in type 2 diabetes. Horm Metab Res. 2004;36:775–781. - PubMed
-
- Gloyn AL, Cummings EA, Edghill EL, Harries LW, Scott R, Costa T, Temple IK, Hattersley AT, Ellard S. Permanent neonatal diabetes due to paternal germline mosaicism for an activating mutation of the KCNJ11 Gene encoding the Kir6.2 subunit of the beta-cell potassium adenosine triphosphate channel. Journal of Clinical Endocrinology & Metabolism. 2004a;89:3932–3935. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
