

Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions
- PMID: 19188606
- PMCID: PMC2634796
- DOI: 10.1073/pnas.0813249106
Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions
Abstract
For comparison of whole-genome (genic + nongenic) sequences, multiple sequence alignment of a few selected genes is not appropriate. One approach is to use an alignment-free method in which feature (or l-mer) frequency profiles (FFP) of whole genomes are used for comparison-a variation of a text or book comparison method, using word frequency profiles. In this approach it is critical to identify the optimal resolution range of l-mers for the given set of genomes compared. The optimum FFP method is applicable for comparing whole genomes or large genomic regions even when there are no common genes with high homology. We outline the method in 3 stages: (i) We first show how the optimal resolution range can be determined with English books which have been transformed into long character strings by removing all punctuation and spaces. (ii) Next, we test the robustness of the optimized FFP method at the nucleotide level, using a mutation model with a wide range of base substitutions and rearrangements. (iii) Finally, to illustrate the utility of the method, phylogenies are reconstructed from concatenated mammalian intronic genomes; the FFP derived intronic genome topologies for each l within the optimal range are all very similar. The topology agrees with the established mammalian phylogeny revealing that intron regions contain a similar level of phylogenic signal as do coding regions.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
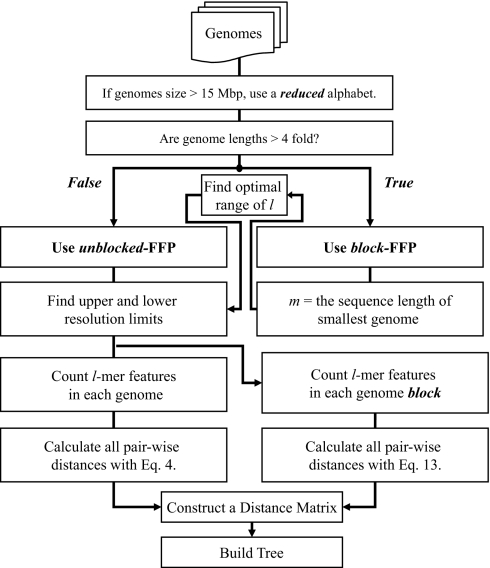
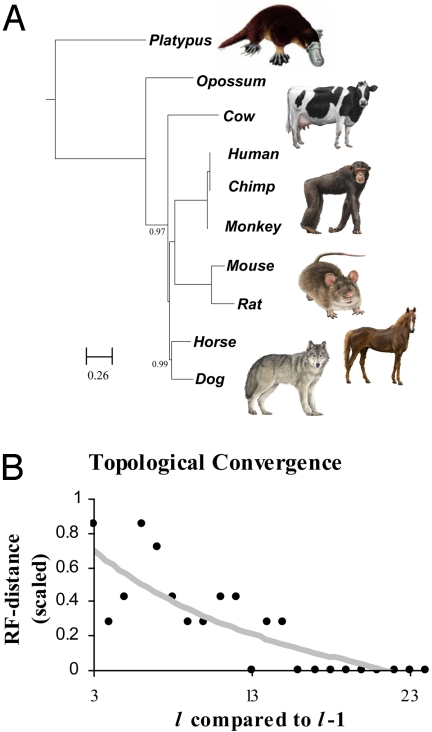
References
-
- Venter JC, et al. The sequence of the human genome. Science. 2001;291:1305–1350.
-
- Deerwester S, et al. Indexing by latent semantic analysis. J Am Soc Inform Sci. 1988;41:391–407.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
