

Loss of pleckstrin defines a novel pathway for PKC-mediated exocytosis
- PMID: 19190246
- PMCID: PMC2668855
- DOI: 10.1182/blood-2008-09-178913
Loss of pleckstrin defines a novel pathway for PKC-mediated exocytosis
Abstract
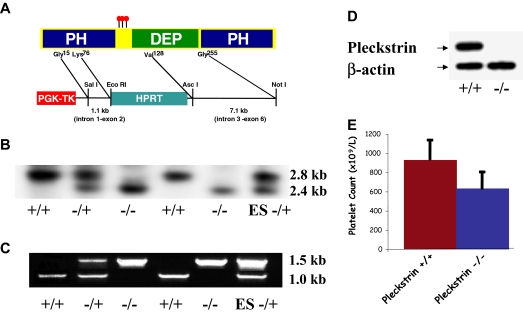
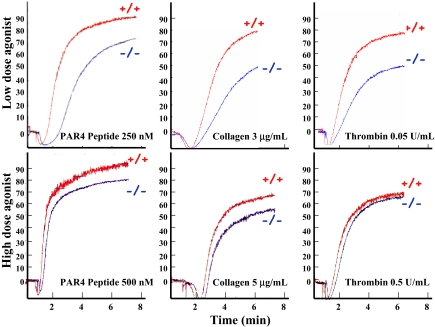
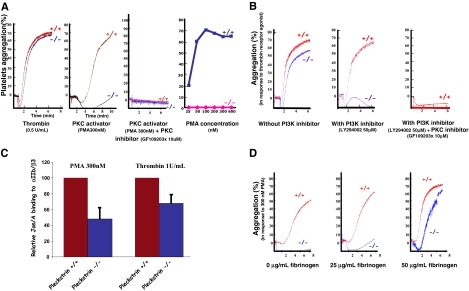
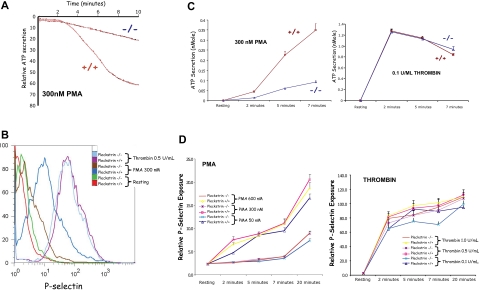
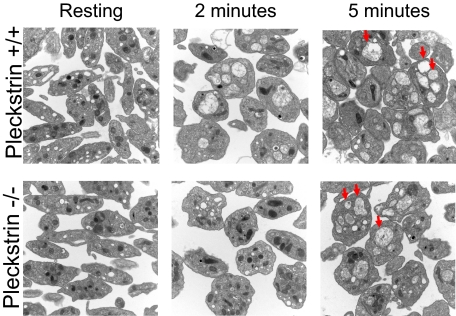
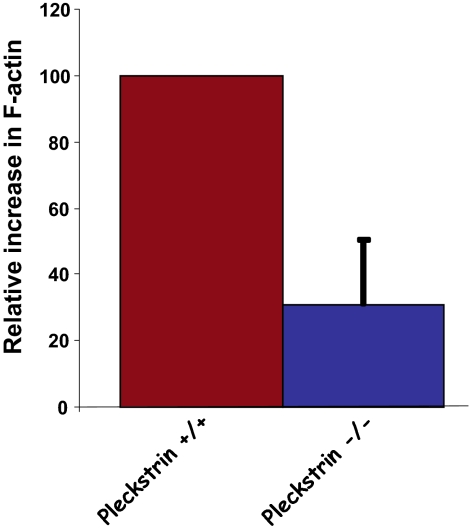
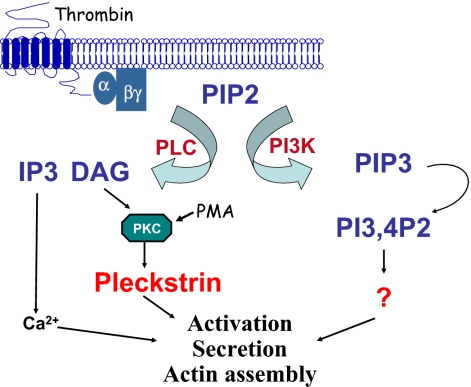
Pleckstrin, the platelet and leukocyte C kinase substrate, is a prominent substrate of PKC in platelets, monocytes, macrophages, lymphocytes, and granulocytes. Pleckstrin accounts for 1% of the total protein in these cells, but it is best known for containing the 2 prototypic Pleckstrin homology, or PH, domains. Overexpressed pleckstrin can affect polyphosphoinositide second messenger-based signaling events; however, its true in vivo role has been unknown. Here, we describe mice containing a null mutation within the pleckstrin gene. Platelets lacking pleckstrin exhibit a marked defect in exocytosis of delta and alpha granules, alphaIIbbeta3 activation, actin assembly, and aggregation after exposure to the PKC stimulant, PMA. Pleckstrin-null platelets aggregate normally in response to thrombin, but they fail to aggregate in response to thrombin in the presence of PI3K inhibitors, suggesting that a PI3K-dependent signaling pathway compensates for the loss of pleckstrin. Although pleckstrin-null platelets merged their granules in response to stimulation of PKC, they failed to empty their contents into the open canalicular system. This might be attributable to impaired actin assembly present in cells lacking pleckstrin. These data show that pleckstrin regulates the fusion of granules to the cell membrane and is an essential component of PKC-mediated exocytosis.
Figures
References
-
- Imaoka T, Lynham JA, Haslam RJ. Purification and characterization of the 47,000-dalton protein phosphorylated during degranulation of human platelets. J Biol Chem. 1983;258:11404–11414. - PubMed
-
- Connolly TM, Lawing WJJ, Majerus PW. Protein kinase C phosphorylates human platelet inositol trisphosphate 5′-phosphomonoesterase, increasing the phosphatase activity. Cell. 1986;46:951–958. - PubMed
-
- Tyers M, Haslam RJ, Rachubinski RA, Harley CB. Molecular analysis of pleckstrin: the major protein kinase C substrate of platelets. J Cell Biochem. 1989;40:133–145. - PubMed
-
- Tyers M, Rachubinski RA, Stewart MI, et al. Molecular cloning and expression of the major protein kinase C substrate of platelets. Nature. 1988;333:470–473. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
