

Ca(2+) permeable AMPA receptor induced long-term potentiation requires PI3/MAP kinases but not Ca/CaM-dependent kinase II
- PMID: 19190753
- PMCID: PMC2629531
- DOI: 10.1371/journal.pone.0004339
Ca(2+) permeable AMPA receptor induced long-term potentiation requires PI3/MAP kinases but not Ca/CaM-dependent kinase II
Abstract
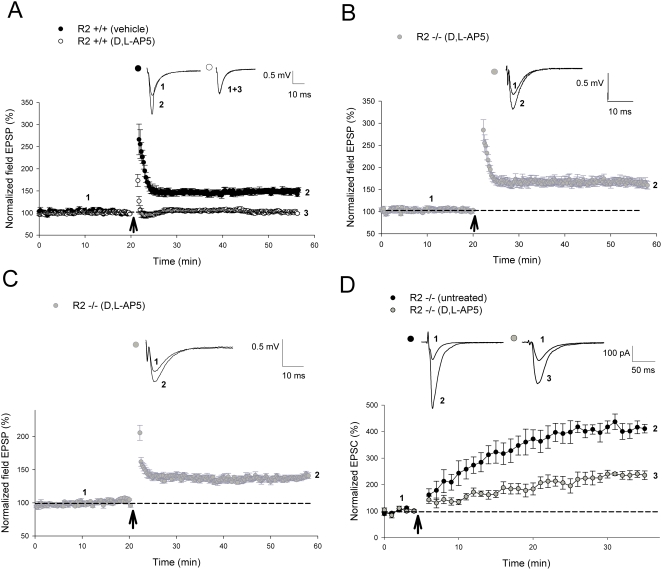
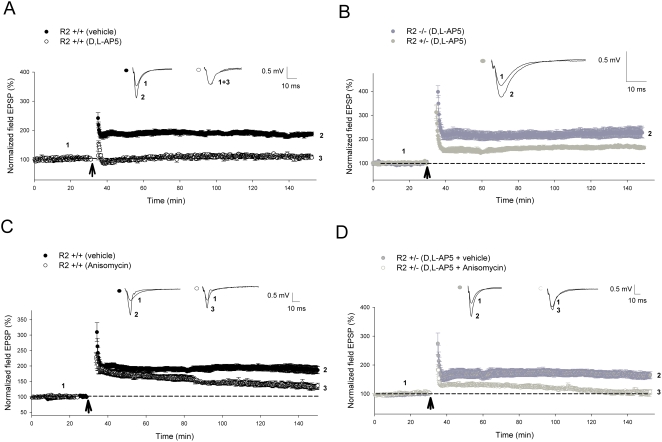
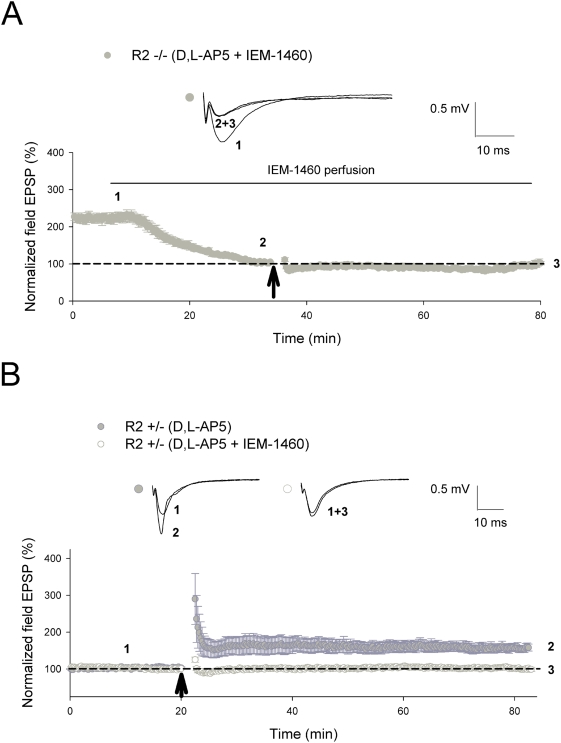
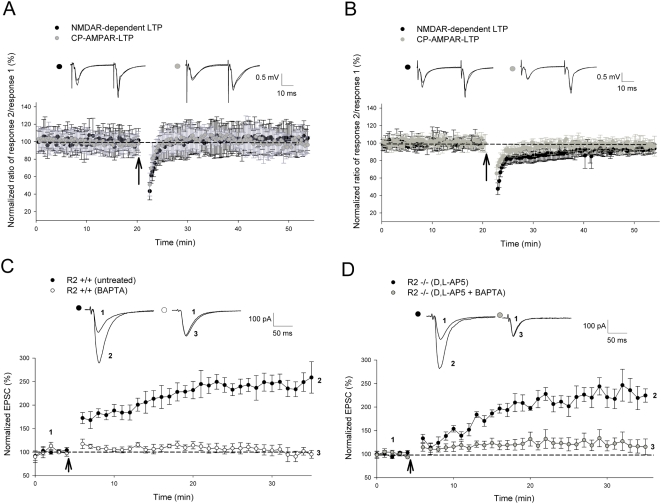
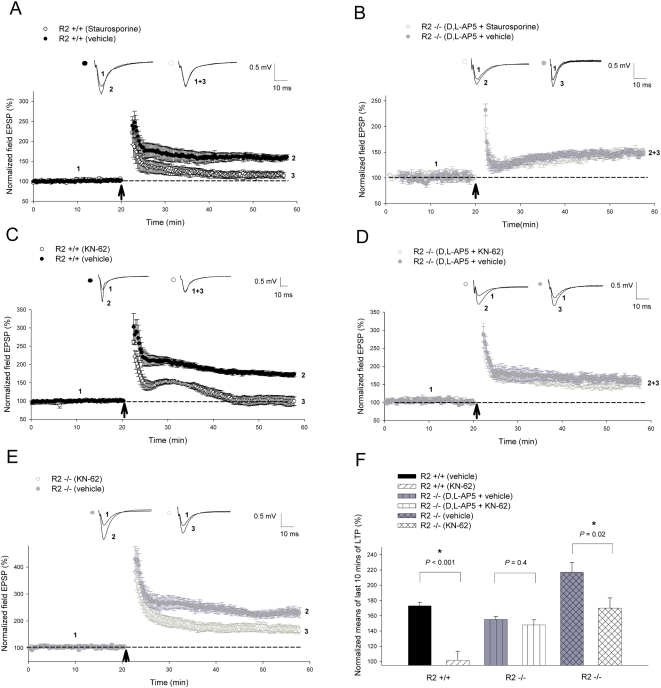
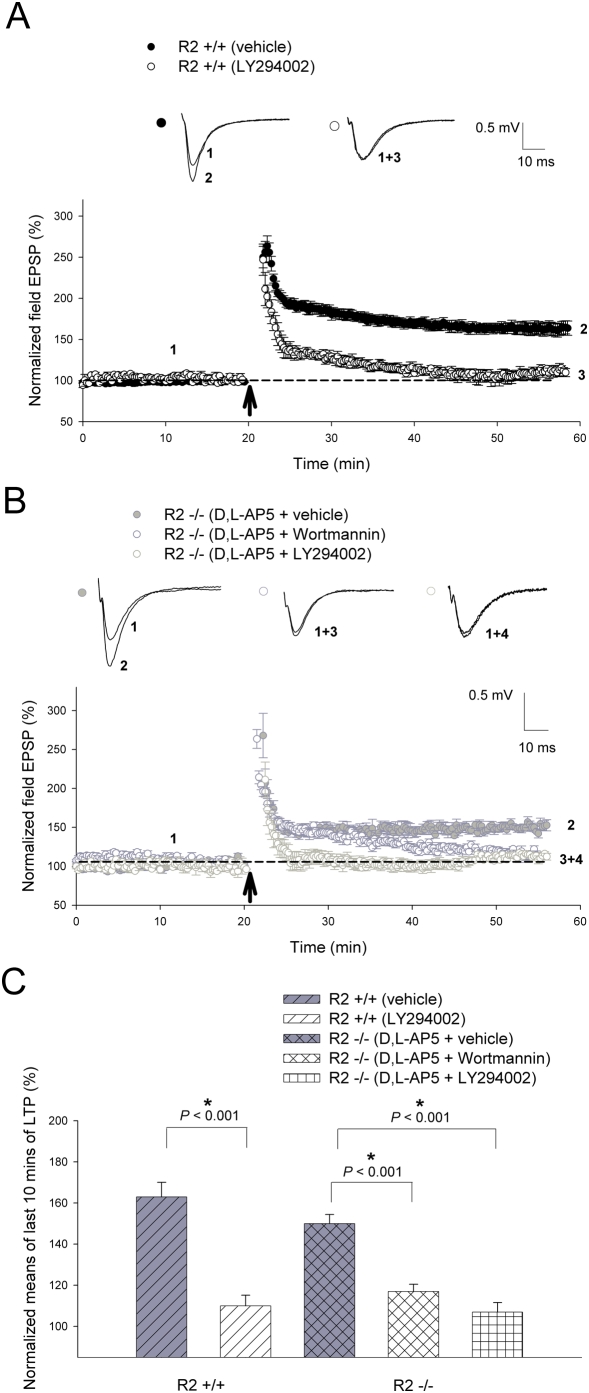
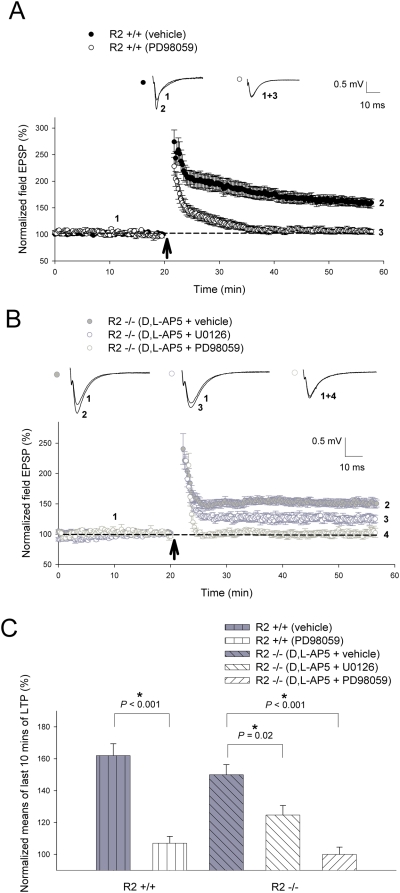
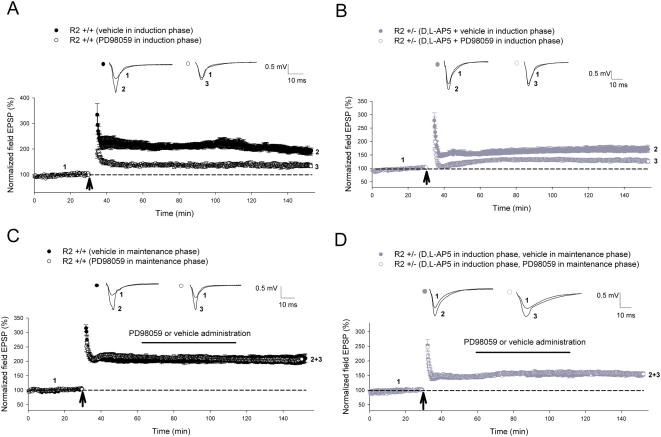
Ca(2+) influx via GluR2-lacking Ca(2+)-permeable AMPA glutamate receptors (CP-AMPARs) can trigger changes in synaptic efficacy in both interneurons and principle neurons, but the underlying mechanisms remain unknown. We took advantage of genetically altered mice with no or reduced GluR2, thus allowing the expression of synaptic CP-AMPARs, to investigate the molecular signaling process during CP-AMPAR-induced synaptic plasticity at CA1 synapses in the hippocampus. Utilizing electrophysiological techniques, we demonstrated that these receptors were capable of inducing numerous forms of long-term potentiation (referred to as CP-AMPAR dependent LTP) through a number of different induction protocols, including high-frequency stimulation (HFS) and theta-burst stimulation (TBS). This included a previously undemonstrated form of protein-synthesis dependent late-LTP (L-LTP) at CA1 synapses that is NMDA-receptor independent. This form of plasticity was completely blocked by the selective CP-AMPAR inhibitor IEM-1460, and found to be dependent on postsynaptic Ca(2+) ions through calcium chelator (BAPTA) studies. Surprisingly, Ca/CaM-dependent kinase II (CaMKII), the key protein kinase that is indispensable for NMDA-receptor dependent LTP at CA1 synapses appeared to be not required for the induction of CP-AMPAR dependent LTP due to the lack of effect of two separate pharmacological inhibitors (KN-62 and staurosporine) on this form of potentiation. Both KN-62 and staurosporine strongly inhibited NMDA-receptor dependent LTP in control studies. In contrast, inhibitors for PI3-kinase (LY294002 and wortmannin) or the MAPK cascade (PD98059 and U0126) significantly attenuated this CP-AMPAR-dependent LTP. Similarly, postsynaptic infusion of tetanus toxin (TeTx) light chain, an inhibitor of exocytosis, also had a significant inhibitory effect on this form of LTP. These results suggest that distinct synaptic signaling underlies GluR2-lacking CP-AMPAR-dependent LTP, and reinforces the recent notions that CP-AMPARs are important facilitators of synaptic plasticity in the brain.
Conflict of interest statement
Figures
References
-
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26. - PubMed
-
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–88. - PubMed
-
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–79. - PubMed
-
- Hestrin S. Different glutamate receptor channels mediate fast excitatory synaptic currents in inhibitory and excitatory cortical neurons. Neuron. 1993;11:1083–91. - PubMed
-
- Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H. Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron. 1994;12:1281–9. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
