

Characterization of pseudorabies virus (PrV) cleavage-encapsidation proteins and functional complementation of PrV pUL32 by the homologous protein of herpes simplex virus type 1
- PMID: 19193798
- PMCID: PMC2663260
- DOI: 10.1128/JVI.02636-08
Characterization of pseudorabies virus (PrV) cleavage-encapsidation proteins and functional complementation of PrV pUL32 by the homologous protein of herpes simplex virus type 1
Abstract
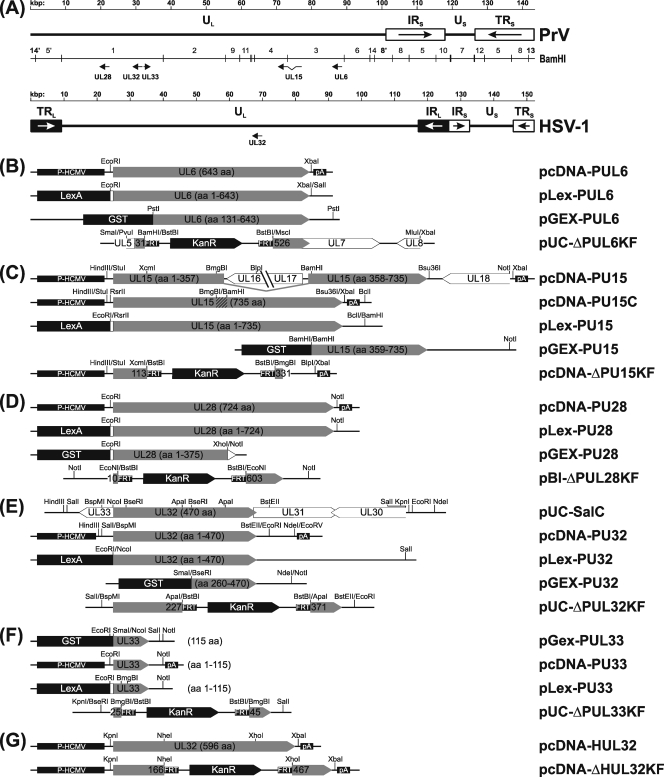
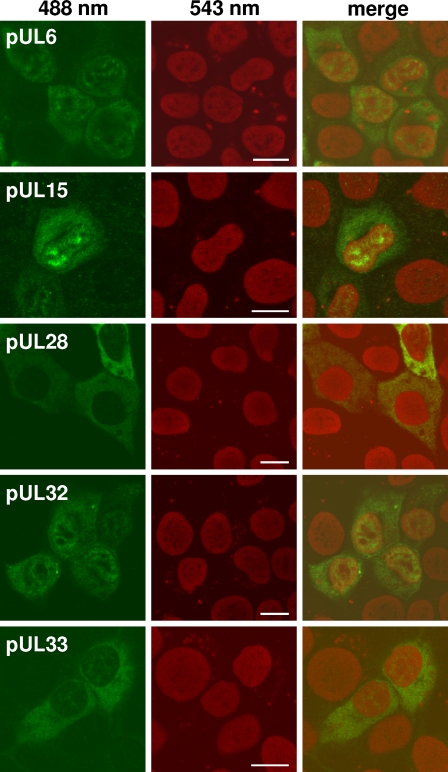
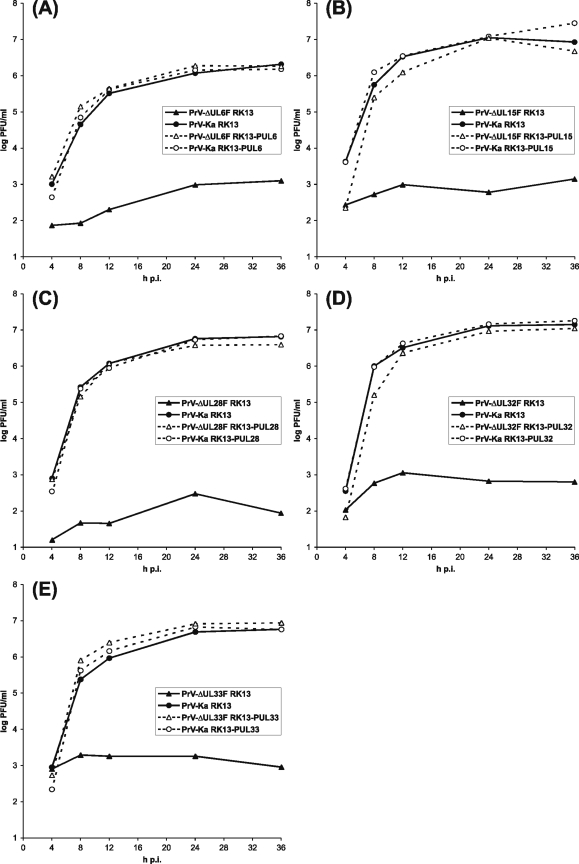
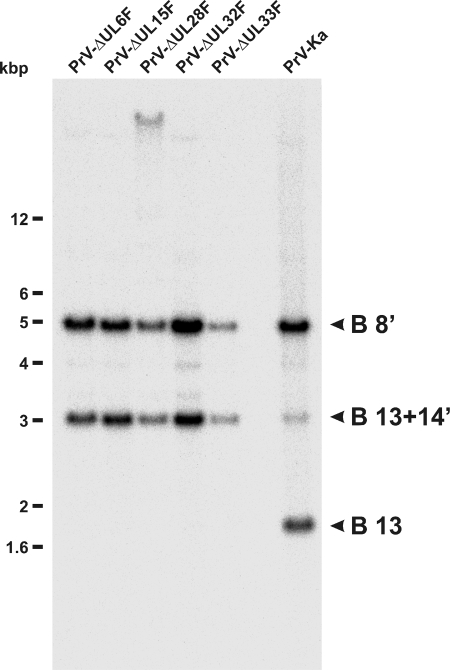
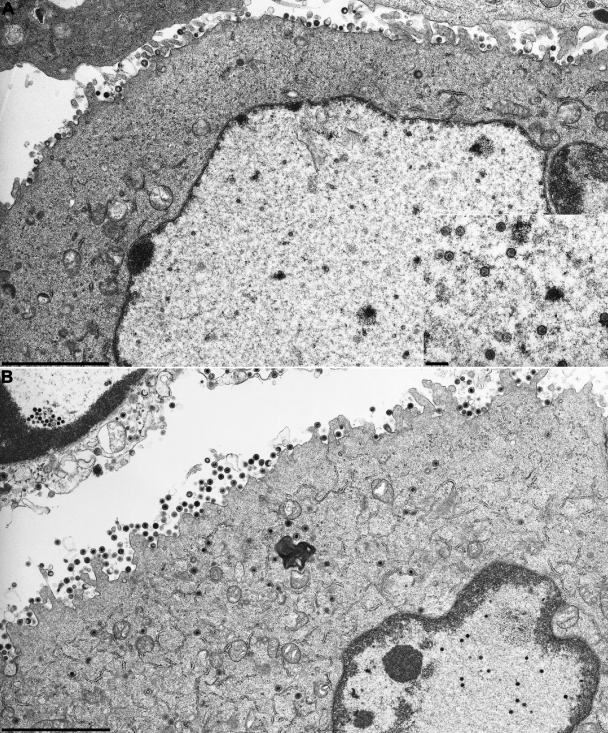
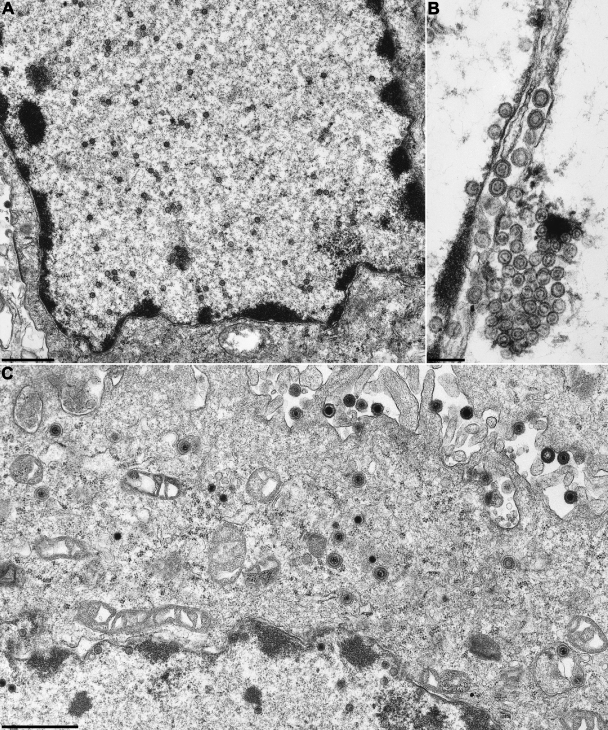
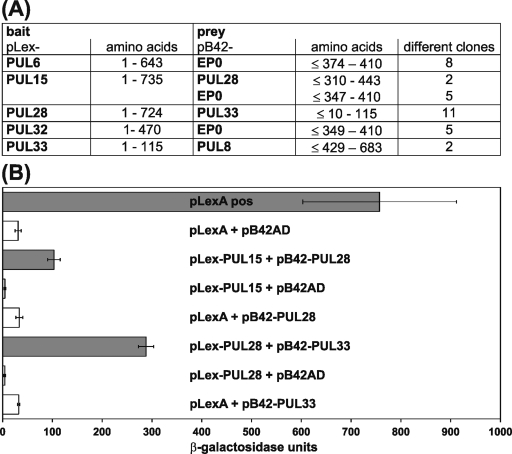
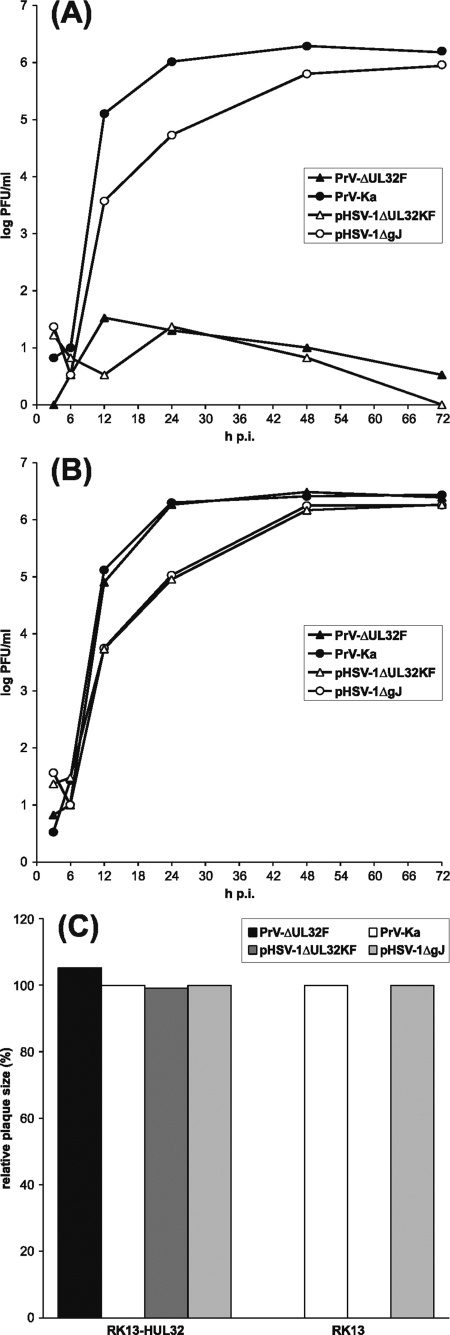
Cleavage and encapsidation of newly replicated herpes simplex virus type 1 (HSV-1) DNA requires several essential viral gene products that are conserved in sequence within the Herpesviridae. However, conservation of function has not been analyzed in greater detail. For functional characterization of the UL6, UL15, UL28, UL32, and UL33 gene products of pseudorabies virus (PrV), the respective deletion mutants were generated by mutagenesis of the virus genome cloned as a bacterial artificial chromosome (BAC) in Escherichia coli and propagated in transgenic rabbit kidney cells lines expressing the deleted genes. Neither of the PrV mutants was able to produce plaques or infectious progeny in noncomplementing cells. DNA analyses revealed that the viral genomes were replicated but not cleaved into monomers. By electron microscopy, only scaffold-containing immature but not DNA-containing mature capsids were detected in the nuclei of noncomplementing cells infected with either of the mutants. Remarkably, primary envelopment of empty capsids at the nuclear membrane occasionally occurred, and enveloped tegument-containing light particles were formed in the cytoplasm and released into the extracellular space. Immunofluorescence analyses with monospecific antisera of cells transfected with the respective expression plasmids indicated that pUL6, pUL15, and pUL32 were able to enter the nucleus. In contrast, pUL28 and pUL33 were predominantly found in the cytoplasm. Only pUL6 could be unequivocally identified and localized in PrV-infected cells and in purified virions, whereas the low abundance or immunogenicity of the other proteins hampered similar studies. Yeast two-hybrid analyses revealed physical interactions between the PrV pUL15, pUL28, and pUL33 proteins, indicating that, as in HSV-1, a tripartite protein complex might catalyze cleavage and encapsidation of viral DNA. Whereas the pUL6 protein is supposed to form the portal for DNA entry into the capsid, the precise role of the UL32 gene product during this process remains to be elucidated. Interestingly, the defect of UL32-negative PrV could be completely corrected in trans by the homologous protein of HSV-1, demonstrating similar functions. However, trans-complementation of UL32-negative HSV-1 by the PrV protein was not observed.
Figures
Similar articles
-
The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions.J Virol. 2002 Jul;76(13):6729-42. doi: 10.1128/jvi.76.13.6729-6742.2002. J Virol. 2002. PMID: 12050386 Free PMC article.
-
The capsid-associated UL25 protein of the alphaherpesvirus pseudorabies virus is nonessential for cleavage and encapsidation of genomic DNA but is required for nuclear egress of capsids.J Virol. 2006 Jul;80(13):6235-46. doi: 10.1128/JVI.02662-05. J Virol. 2006. PMID: 16775311 Free PMC article.
-
Role of the Herpes Simplex Virus CVSC Proteins at the Capsid Portal Vertex.J Virol. 2020 Nov 23;94(24):e01534-20. doi: 10.1128/JVI.01534-20. Print 2020 Nov 23. J Virol. 2020. PMID: 32967953 Free PMC article.
-
Herpesvirus Capsid Assembly and DNA Packaging.Adv Anat Embryol Cell Biol. 2017;223:119-142. doi: 10.1007/978-3-319-53168-7_6. Adv Anat Embryol Cell Biol. 2017. PMID: 28528442 Free PMC article. Review.
-
Tegument Assembly and Secondary Envelopment of Alphaherpesviruses.Viruses. 2015 Sep 18;7(9):5084-114. doi: 10.3390/v7092861. Viruses. 2015. PMID: 26393641 Free PMC article. Review.
Cited by
-
Changes in subcellular localization reveal interactions between human cytomegalovirus terminase subunits.Virol J. 2012 Dec 21;9:315. doi: 10.1186/1743-422X-9-315. Virol J. 2012. PMID: 23259714 Free PMC article.
-
Characterization of pseudorabies virus transcriptome by Illumina sequencing.BMC Microbiol. 2015 Jul 1;15:130. doi: 10.1186/s12866-015-0470-0. BMC Microbiol. 2015. PMID: 26129912 Free PMC article.
-
The Apical Region of the Herpes Simplex Virus Major Capsid Protein Promotes Capsid Maturation.J Virol. 2018 Aug 29;92(18):e00821-18. doi: 10.1128/JVI.00821-18. Print 2018 Sep 15. J Virol. 2018. PMID: 29976665 Free PMC article.
-
Characterization of the Varicella-zoster virus ORF25 gene product: pORF25 interacts with multiple DNA encapsidation proteins.Virus Res. 2009 Sep;144(1-2):58-64. doi: 10.1016/j.virusres.2009.03.019. Epub 2009 Apr 7. Virus Res. 2009. PMID: 19720242 Free PMC article.
-
The effects of viral load on pseudorabies virus gene expression.BMC Microbiol. 2010 Dec 6;10:311. doi: 10.1186/1471-2180-10-311. BMC Microbiol. 2010. PMID: 21134263 Free PMC article.
References
-
- al-Kobaisi, M. F., F. J. Rixon, I. McDougall, and V. G. Preston. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180380-388. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
