

Clinical heterogeneity of duchenne muscular dystrophy (DMD): definition of sub-phenotypes and predictive criteria by long-term follow-up
- PMID: 19194511
- PMCID: PMC2633042
- DOI: 10.1371/journal.pone.0004347
Clinical heterogeneity of duchenne muscular dystrophy (DMD): definition of sub-phenotypes and predictive criteria by long-term follow-up
Abstract
Background: To explore clinical heterogeneity of Duchenne muscular dystrophy (DMD), viewed as a major obstacle to the interpretation of therapeutic trials
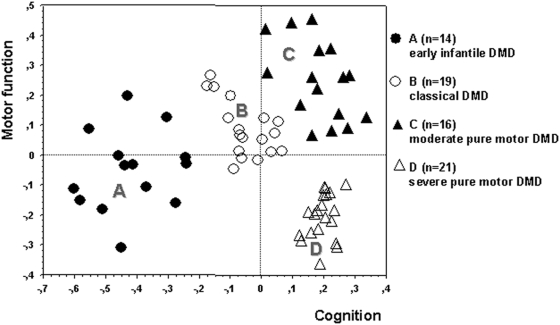
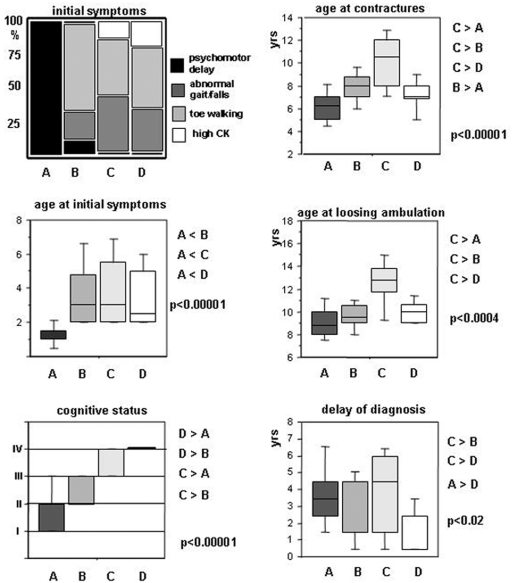
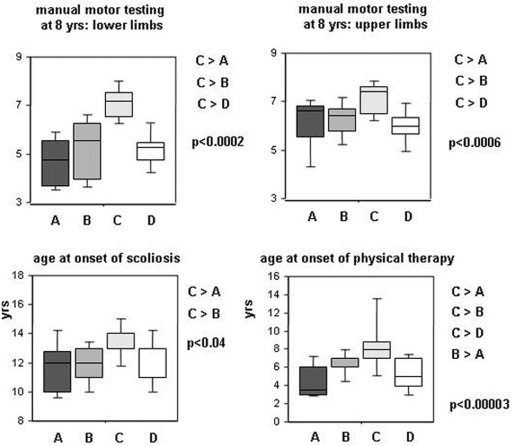
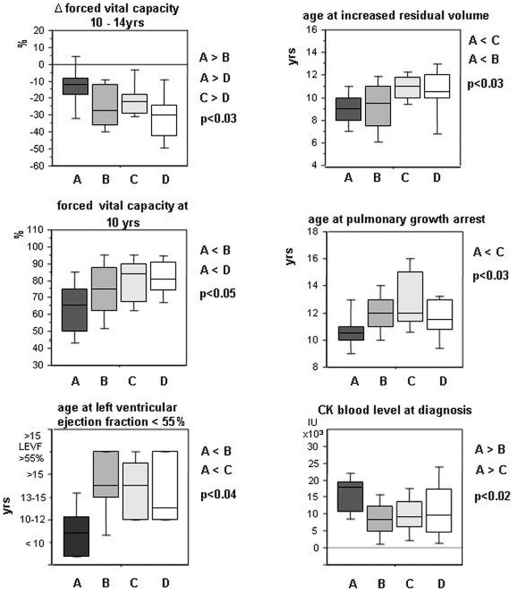
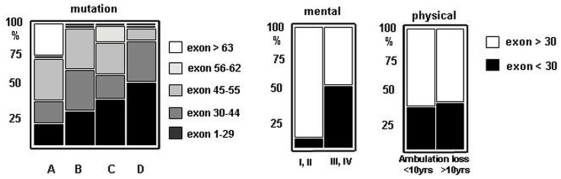
Methodology/principal findings: A retrospective single institution long-term follow-up study was carried out in DMD patients with both complete lack of muscle dystrophin and genotyping. An exploratory series (series 1) was used to assess phenotypic heterogeneity and to identify early criteria predicting future outcome; it included 75 consecutive steroid-free patients, longitudinally evaluated for motor, respiratory, cardiac and cognitive functions (median follow-up: 10.5 yrs). A validation series (series 2) was used to test robustness of the selected predictive criteria; it included 34 more routinely evaluated patients (age>12 yrs). Multivariate analysis of series 1 classified 70/75 patients into 4 clusters with distinctive intellectual and motor outcomes: A (early infantile DMD, 20%): severe intellectual and motor outcomes; B (classical DMD, 28%): intermediate intellectual and poor motor outcome; C (moderate pure motor DMD, 22%): normal intelligence and delayed motor impairment; and D (severe pure motor DMD, 30%): normal intelligence and poor motor outcome. Group A patients had the most severe respiratory and cardiac involvement. Frequency of mutations upstream to exon 30 increased from group A to D, but genotype/phenotype correlations were restricted to cognition (IQ>71: OR 7.7, 95%CI 1.6-20.4, p<0.003). Diagnostic accuracy tests showed that combination of "clinical onset <2 yrs" with "mental retardation" reliably assigned patients to group A (sensitivity 0.93, specificity 0.98). Combination of "lower limb MMT score>6 at 8 yrs" with "normal or borderline mental status" reliably assigned patients to group C (sensitivity: 1, specificity: 0.94). These criteria were also predictive of "early infantile DMD" and "moderate pure motor DMD" in series 2.
Conclusions/significance: DMD can be divided into 4 sub-phenotypes differing by severity of muscle and brain dysfunction. Simple early criteria can be used to include patients with similar outcomes in future therapeutic trials.
Conflict of interest statement
Figures
References
-
- Brooke MH, Fenichel GM, Griggs RC, Mendell JR, Moxley R, et al. Duchenne muscular dystrophy: patterns of clinical progression and effects of supportive therapy. Neurology. 1989;39:475–481. - PubMed
-
- Brooke MH, Fenichel GM, Griggs RC, Schumate JB, Pellegrino RJ. Clinical investigation in Duchenne Dystrophy:Determination of the power of therapeutic trials based on natural history. Muscle Nerve. 1983;6:91–103. - PubMed
-
- Mendell JR, Province MA, Griggs RC, Brooke MH, Fenichel GM, et al. Clinical investigation of Duchenne muscular dystrophy. A methodology for therapeutic trials based on natural history controls. Arch Neurol. 1987;44:808–811. - PubMed
-
- Sifringer M, Uhlenberg B, Lammel S, Hanke R, Neumann B, et al. Identification of transcripts from a subtraction library which might be responsible for the mild phenotype in an intrafamilially variable course of Duchenne muscular dystrophy. Hum Genet. 2004;114:149–156. - PubMed
-
- Bushby KM, Apleton R, Anderson LV, Welsch JL, Kelly P, et al. Deletion status and intellectual impairment in Duchenne muscular dystrophy. Dev Med Child Neurol. 1995;37:260–269. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
