

Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy
- PMID: 19197364
- PMCID: PMC2631153
- DOI: 10.1371/journal.pgen.1000372
Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy
Abstract
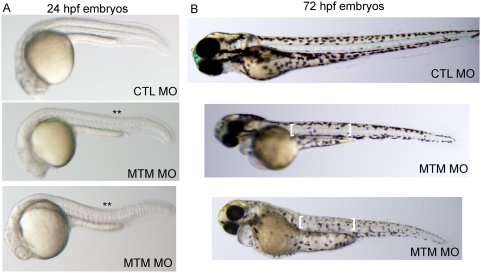
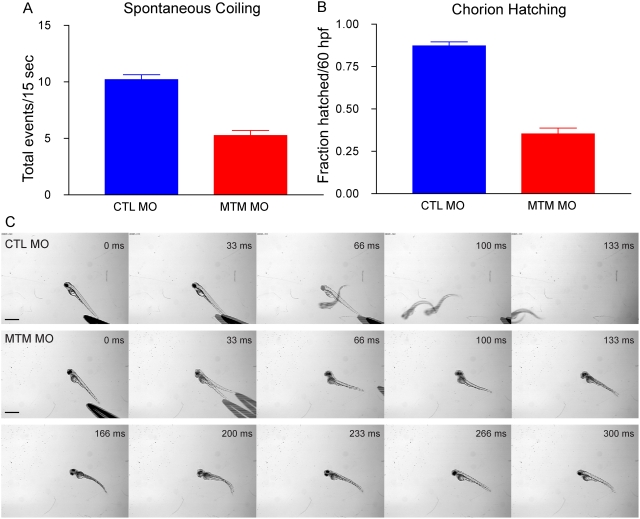
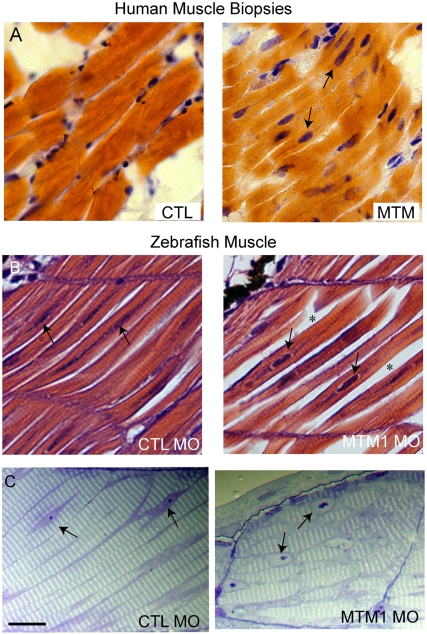
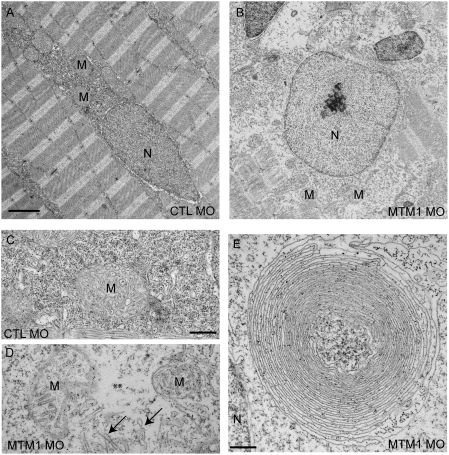
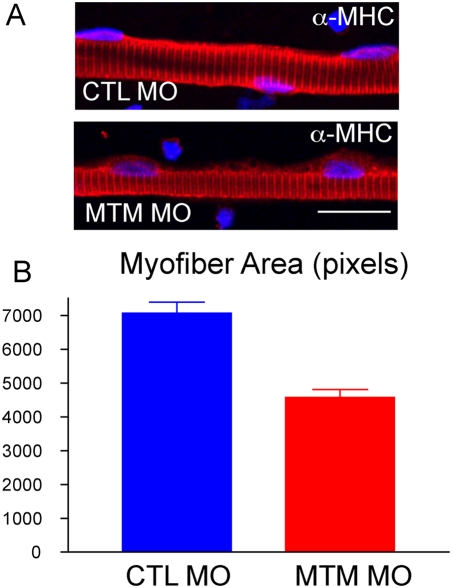
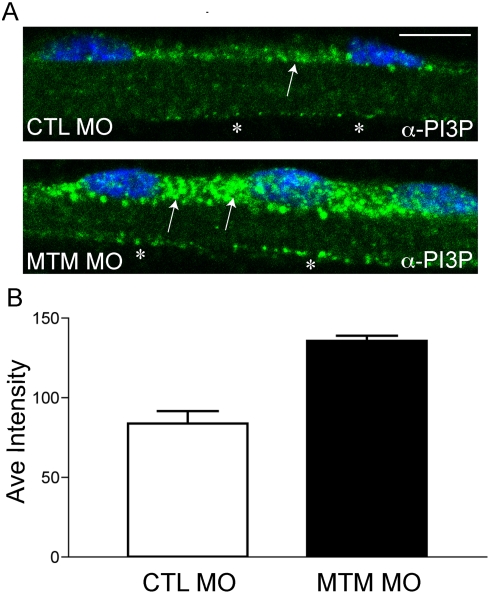
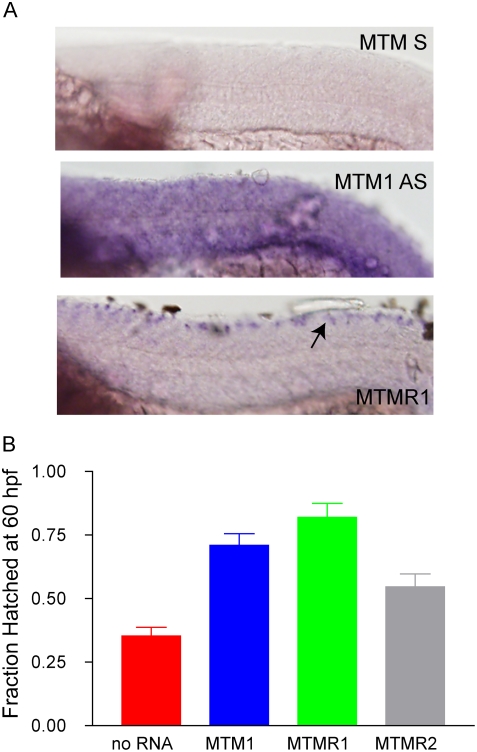
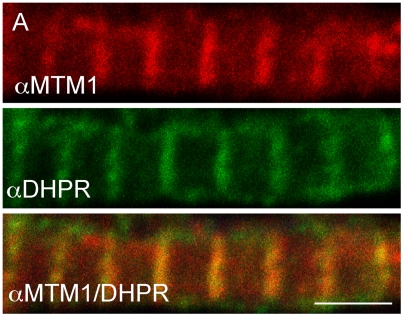
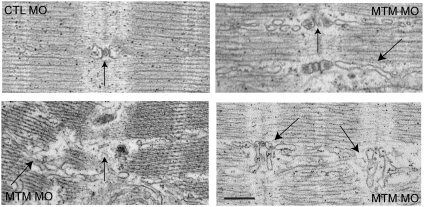
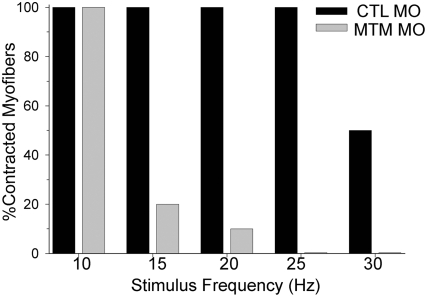
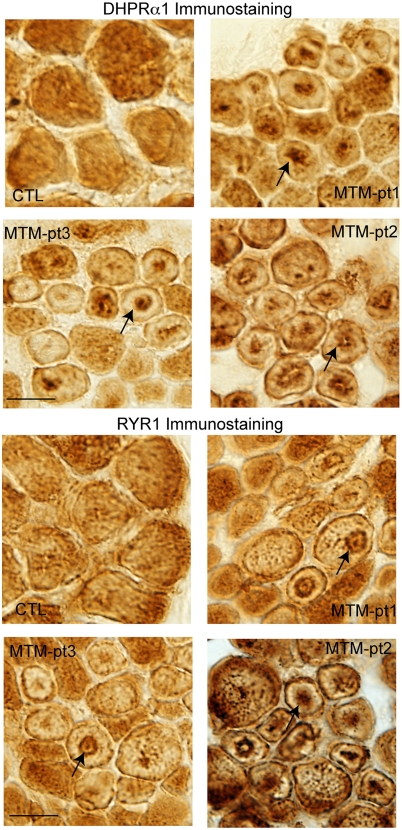
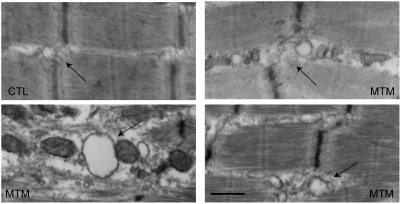
Myotubularin is a lipid phosphatase implicated in endosomal trafficking in vitro, but with an unknown function in vivo. Mutations in myotubularin cause myotubular myopathy, a devastating congenital myopathy with unclear pathogenesis and no current therapies. Myotubular myopathy was the first described of a growing list of conditions caused by mutations in proteins implicated in membrane trafficking. To advance the understanding of myotubularin function and disease pathogenesis, we have created a zebrafish model of myotubular myopathy using morpholino antisense technology. Zebrafish with reduced levels of myotubularin have significantly impaired motor function and obvious histopathologic changes in their muscle. These changes include abnormally shaped and positioned nuclei and myofiber hypotrophy. These findings are consistent with those observed in the human disease. We demonstrate for the first time that myotubularin functions to regulate PI3P levels in a vertebrate in vivo, and that homologous myotubularin-related proteins can functionally compensate for the loss of myotubularin. Finally, we identify abnormalities in the tubulo-reticular network in muscle from myotubularin zebrafish morphants and correlate these changes with abnormalities in T-tubule organization in biopsies from patients with myotubular myopathy. In all, we have generated a new model of myotubular myopathy and employed this model to uncover a novel function for myotubularin and a new pathomechanism for the human disease that may explain the weakness associated with the condition (defective excitation-contraction coupling). In addition, our findings of tubuloreticular abnormalities and defective excitation-contraction coupling mechanistically link myotubular myopathy with several other inherited muscle diseases, most notably those due to ryanodine receptor mutations. Based on our findings, we speculate that congenital myopathies, usually considered entities with similar clinical features but very disparate pathomechanisms, may at their root be disorders of calcium homeostasis.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Pierson CR, Tomczak K, Agrawal P, Moghadaszadeh B, Beggs AH. X-linked myotubular and centronuclear myopathies. J Neuropathol Exp Neurol. 2005;64:555–564. - PubMed
-
- Dubowitz V, Sewry CA. Muscle BIopsy: A Practical Approach: Saunders. 2006:626.
-
- Laporte J, Hu LJ, Kretz C, Mandel JL, Kioschis P, et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13:175–182. - PubMed
-
- Clague MJ, Lorenzo O. The myotubularin family of lipid phosphatases. Traffic. 2005;6:1063–1069. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
