

The role of LMNA in adipose: a novel mouse model of lipodystrophy based on the Dunnigan-type familial partial lipodystrophy mutation
- PMID: 19201734
- PMCID: PMC2681389
- DOI: 10.1194/jlr.M800491-JLR200
The role of LMNA in adipose: a novel mouse model of lipodystrophy based on the Dunnigan-type familial partial lipodystrophy mutation
Abstract
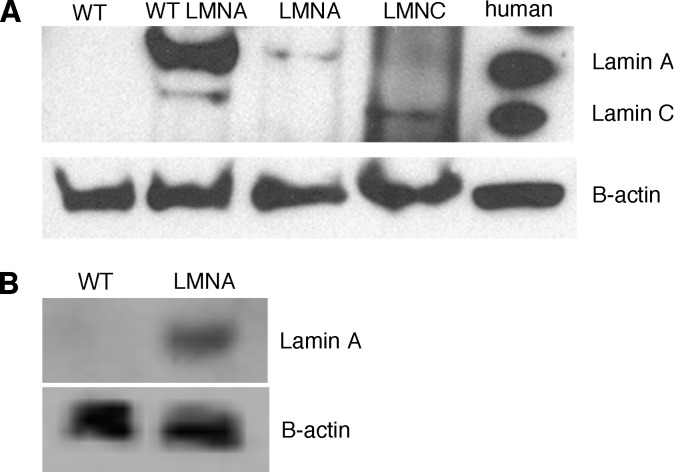
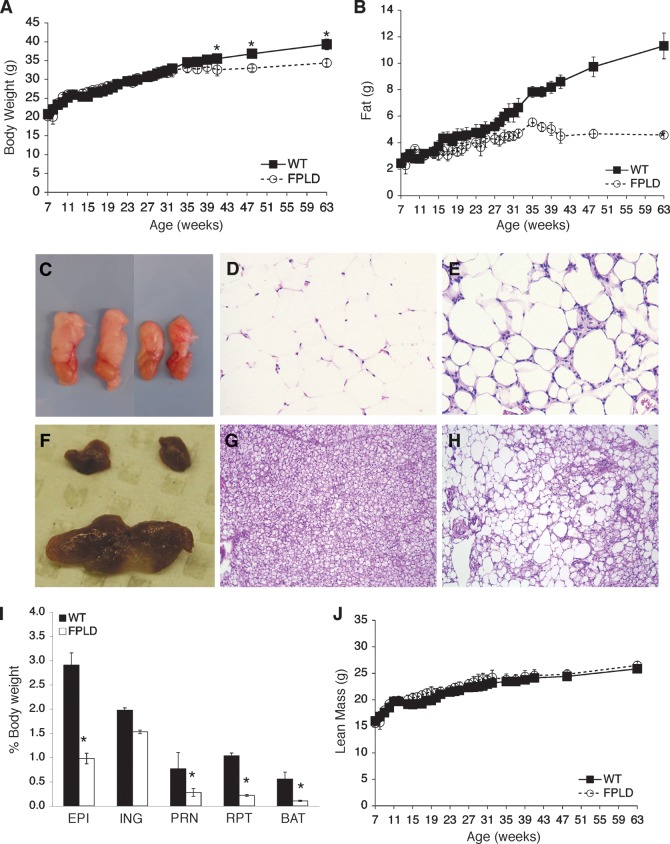
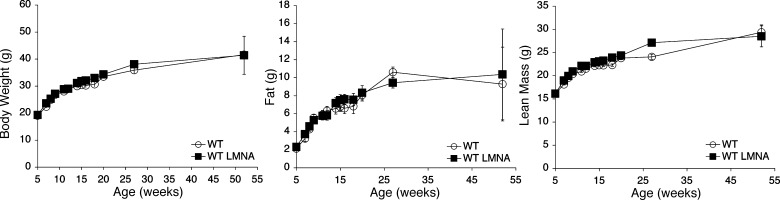
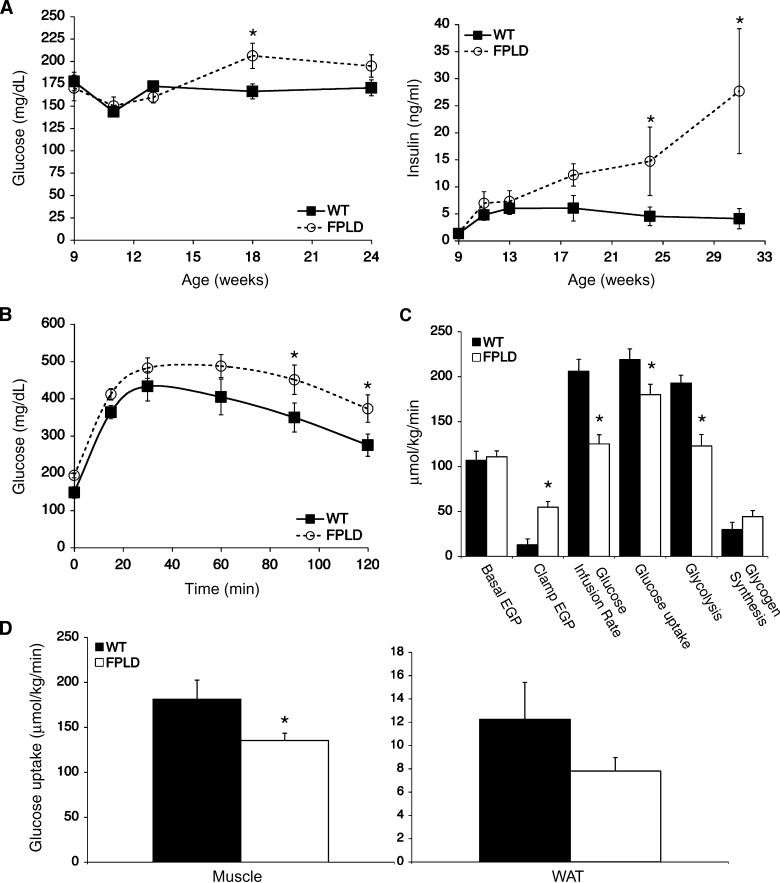
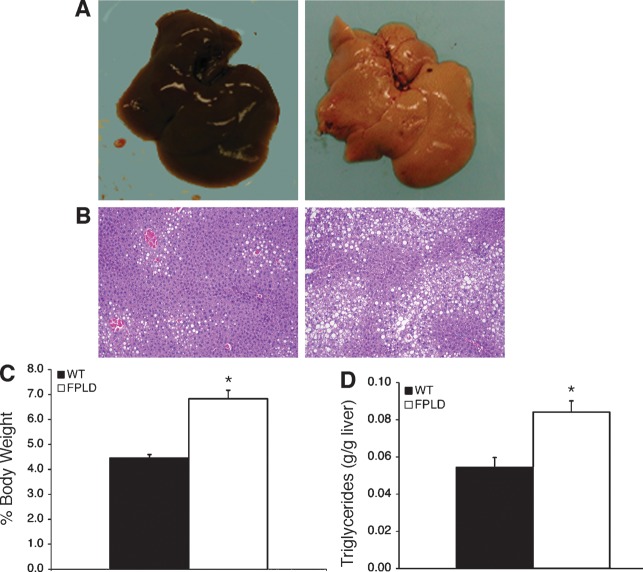
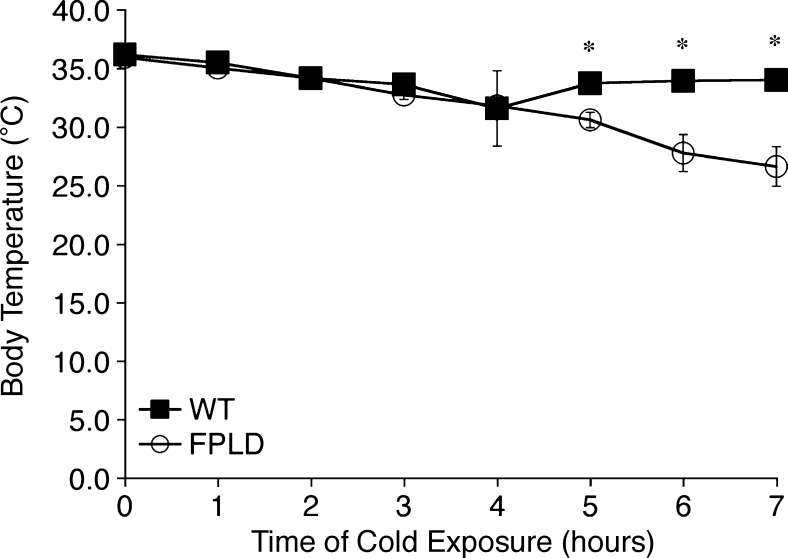
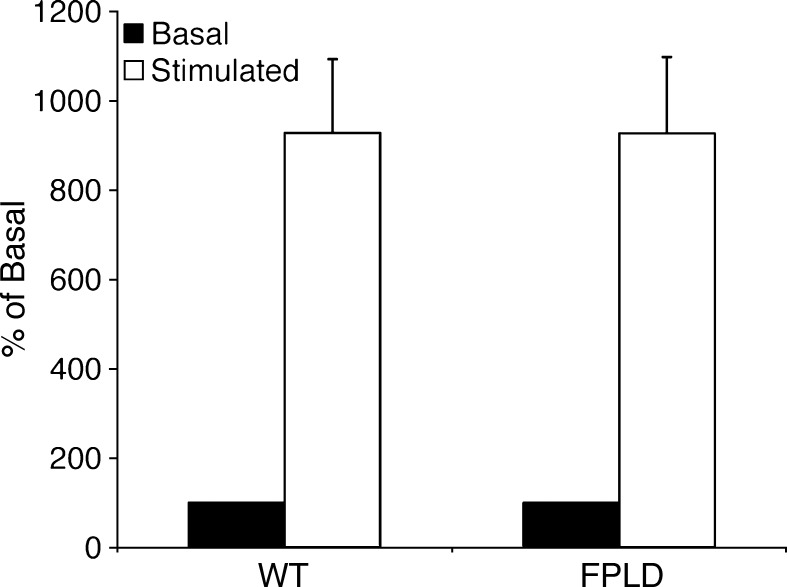
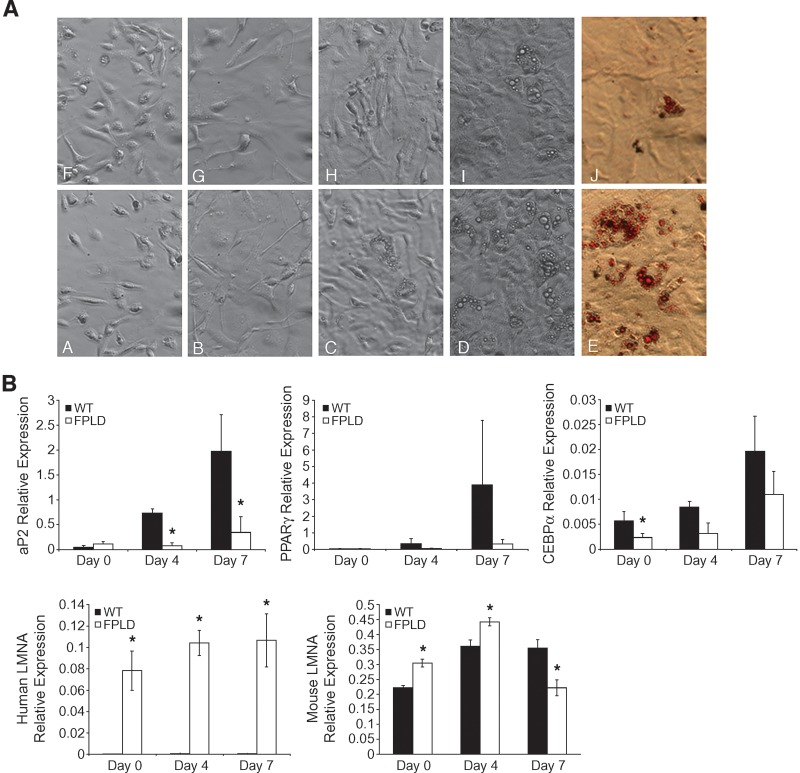
We investigated the role of LMNA in adipose tissue by developing a novel mouse model of lipodystrophy. Transgenic mice were generated that express the LMNA mutation that causes familial partial lipodystrophy of the Dunnigan type (FPLD2). The phenotype observed in FPLD-transgenic mice resembles many of the features of human FPLD2, including lack of fat accumulation, insulin resistance, and enlarged, fatty liver. Similar to the human disease, FPLD-transgenic mice appear to develop normally, but after several weeks they are unable to accumulate fat to the same extent as their wild-type littermates. One poorly understood aspect of lipodystrophies is the mechanism of fat loss. To this end, we have examined the effects of the FPLD2 mutation on fat cell function. Contrary to the current literature, which suggests FPLD2 results in a loss of fat, we found that the key mechanism contributing to the lack of fat accumulation involves not a loss, but an apparent inability of the adipose tissue to renew itself. Specifically, preadipocytes are unable to differentiate into mature and fully functional adipocytes. These findings provide insights not only for the treatment of lipodystrophies, but also for the study of adipogenesis, obesity, and insulin resistance.
Figures
References
-
- Agarwal A. K., and A. Garg. 2006. Genetic basis of lipodystrophies and management of metabolic complications. Annu. Rev. Med. 57 297–311. - PubMed
-
- Rajala M. W., and P. E. Scherer. 2003. Minireview: The adipocyte–at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 144 3765–3773. - PubMed
-
- Cao H., and R. A. Hegele. 2000. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 9 109–112. - PubMed
-
- Jacob K. N., and A. Garg. 2006. Laminopathies: multisystem dystrophy syndromes. Mol. Genet. Metab. 87 289–302. - PubMed
-
- Garg A., R. M. Peshock, and J. L. Fleckenstein. 1999. Adipose tissue distribution pattern in patients with familial partial lipodystrophy (Dunnigan variety). J. Clin. Endocrinol. Metab. 84 170–174. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
