

The death domain of FADD is essential for embryogenesis, lymphocyte development, and proliferation
- PMID: 19203997
- PMCID: PMC2665115
- DOI: 10.1074/jbc.M900249200
The death domain of FADD is essential for embryogenesis, lymphocyte development, and proliferation
Abstract
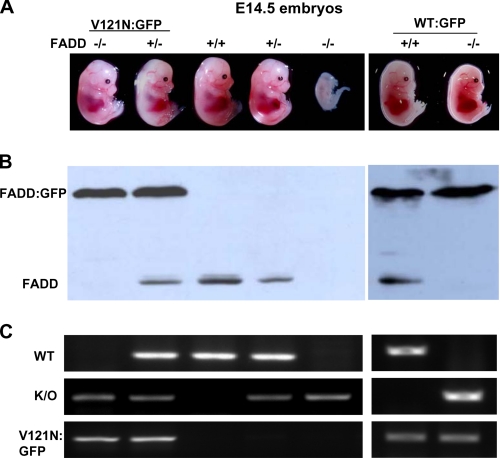
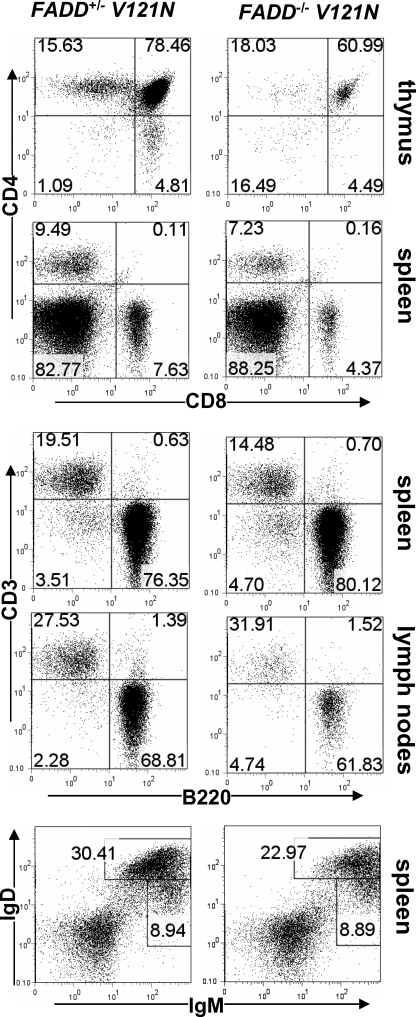
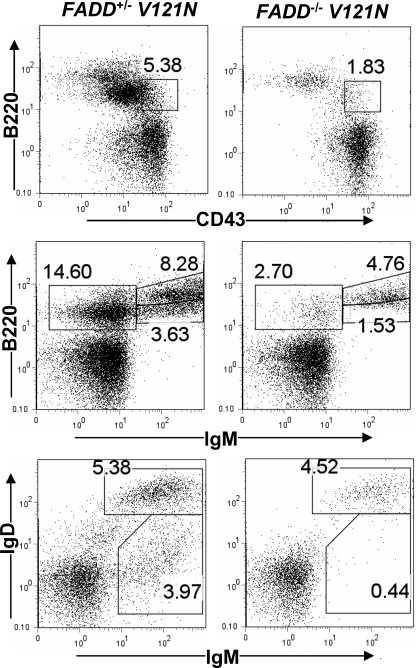
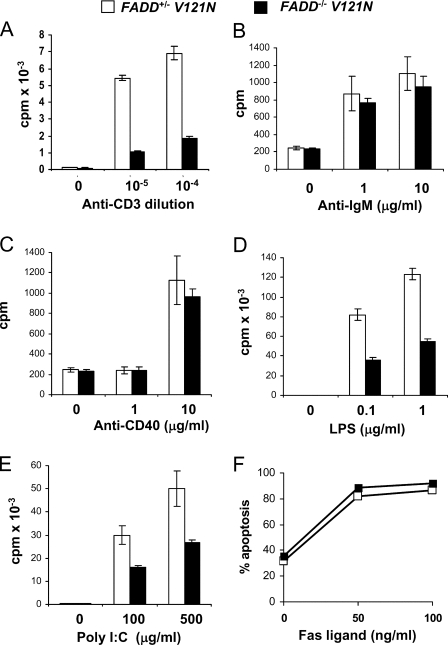
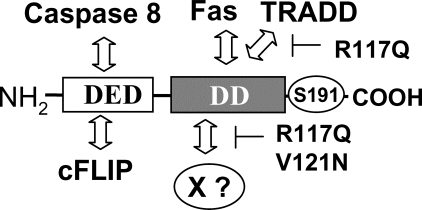
The Fas-associated death domain-containing protein (FADD) is an adaptor for relaying apoptotic signals initiated by death receptors such as Fas. Whereas a lack of death receptors has no effect on mouse development, FADD deficiency results in early embryonic lethality, indicating that FADD has additional functions independent of death receptors. We have previously shown that conditional deletion of FADD not only impairs apoptosis but also leads to defective lymphocyte proliferation. The non-apoptotic signaling mediated by FADD remains poorly understood. Earlier studies have suggested that FADD carboxyl terminal serine phosphorylation likely plays a role in FADD-mediated proliferation signaling in T cells. The FADD death domain is presumably only required for apoptotic signaling, as it interacts with death receptors which are dispensable during embryonic development and lymphocyte proliferation. To test this hypothesis, we have performed mutational analyses of the FADD death domain and identified a mutant, R117Q, which lacks binding to Fas and, thus, is incapable of apoptotic signaling in cell lines. Unexpectedly, this death domain point mutation disrupted mouse embryonic development as shown by in vivo functional reconstitution analyses. Interestingly, a second FADD death domain mutant, V121N, retained normal Fas binding and apoptotic signaling ability but also failed to support mouse development. Furthermore, lymphocyte proliferation responses were impaired by V121N. This reverse genetic study has revealed a previously unappreciated role of the FADD death domain, which likely functions as a molecular switch regulating two distinct signals leading to apoptosis and cell proliferation and is critical for embryogenesis, lymphocyte development, and proliferation.
Figures
Similar articles
-
Structural requirements for signal-induced target binding of FADD determined by functional reconstitution of FADD deficiency.J Biol Chem. 2005 Sep 9;280(36):31360-7. doi: 10.1074/jbc.M504138200. Epub 2005 Jul 11. J Biol Chem. 2005. PMID: 16009710
-
Functional complementation between FADD and RIP1 in embryos and lymphocytes.Nature. 2011 Mar 17;471(7338):373-6. doi: 10.1038/nature09878. Epub 2011 Mar 2. Nature. 2011. PMID: 21368761 Free PMC article.
-
Conditional Fas-associated death domain protein (FADD): GFP knockout mice reveal FADD is dispensable in thymic development but essential in peripheral T cell homeostasis.J Immunol. 2005 Sep 1;175(5):3033-44. doi: 10.4049/jimmunol.175.5.3033. J Immunol. 2005. PMID: 16116191 Free PMC article.
-
The roles of FADD in extrinsic apoptosis and necroptosis.BMB Rep. 2012 Sep;45(9):496-508. doi: 10.5483/bmbrep.2012.45.9.186. BMB Rep. 2012. PMID: 23010170 Review.
-
FADD and its phosphorylation.IUBMB Life. 2004 Jul;56(7):395-401. doi: 10.1080/15216540400008929. IUBMB Life. 2004. PMID: 15545216 Review.
Cited by
-
FADD Phosphorylation Modulates Blood Glucose Levels by Decreasing the Expression of Insulin-Degrading Enzyme.Mol Cells. 2020 Apr 30;43(4):373-383. doi: 10.14348/molcells.2020.2198. Mol Cells. 2020. PMID: 32191993 Free PMC article.
-
The role of death effector domain-containing proteins in acute oxidative cell injury in hepatocytes.Free Radic Biol Med. 2012 May 1;52(9):1911-7. doi: 10.1016/j.freeradbiomed.2012.02.049. Epub 2012 Mar 8. Free Radic Biol Med. 2012. PMID: 22406316 Free PMC article.
-
Developmental checkpoints guarded by regulated necrosis.Cell Mol Life Sci. 2016 Jun;73(11-12):2125-36. doi: 10.1007/s00018-016-2188-z. Epub 2016 Apr 7. Cell Mol Life Sci. 2016. PMID: 27056574 Free PMC article. Review.
-
Increased Expression of Phosphorylated FADD in Anaplastic Large Cell and Other T-Cell Lymphomas.Biomark Insights. 2014 Sep 3;9:77-84. doi: 10.4137/BMI.S16553. eCollection 2014. Biomark Insights. 2014. PMID: 25232277 Free PMC article.
-
Apoptotic cell death in disease-Current understanding of the NCCD 2023.Cell Death Differ. 2023 May;30(5):1097-1154. doi: 10.1038/s41418-023-01153-w. Epub 2023 Apr 26. Cell Death Differ. 2023. PMID: 37100955 Free PMC article. Review.
References
-
- Tartaglia, L. A., Ayres, T. M., Wong, G. H. W., and Goeddel, D. V. (1993) Cell 74 845–853 - PubMed
-
- Itoh, N., and Nagata, S. (1993) J. Biol. Chem. 268 10932–10937 - PubMed
-
- Nagata, S., and Golstein, P. (1995) Science 267 1449–1455 - PubMed
-
- Fisher, G. H., Rosenberg, F. J., Straus, S. E., Dale, J. K., Middleton, L. A., Lin, A. Y., Strober, W., Lenardo, M. J., and Puck, J. M. (1995) Cell 81 935–946 - PubMed
-
- Hill, L. L., Ouhtit, A., Loughlin, S. M., Kripke, M. L., Ananthaswamy, H. N., and Owen-Schaub, L. B. (1999) Science 285 898–900 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
