

c-Jun N-terminal kinase 1 phosphorylates Myt1 to prevent UVA-induced skin cancer
- PMID: 19204086
- PMCID: PMC2663297
- DOI: 10.1128/MCB.01508-08
c-Jun N-terminal kinase 1 phosphorylates Myt1 to prevent UVA-induced skin cancer
Abstract
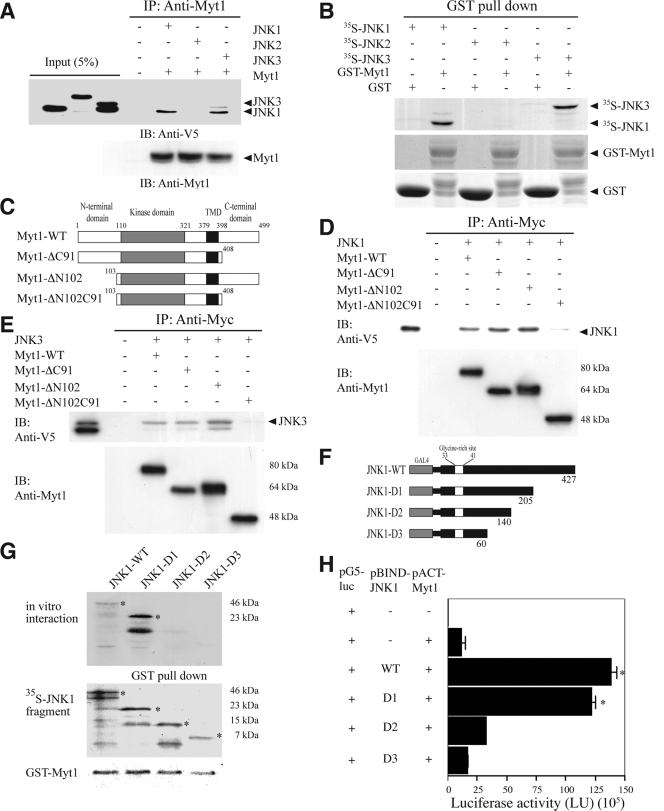
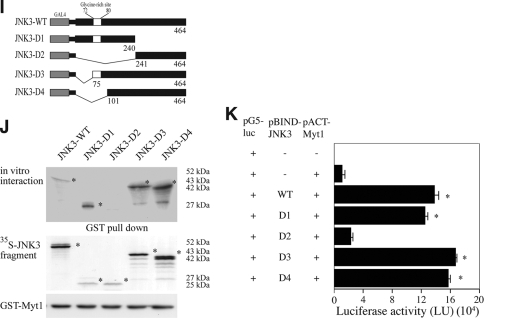
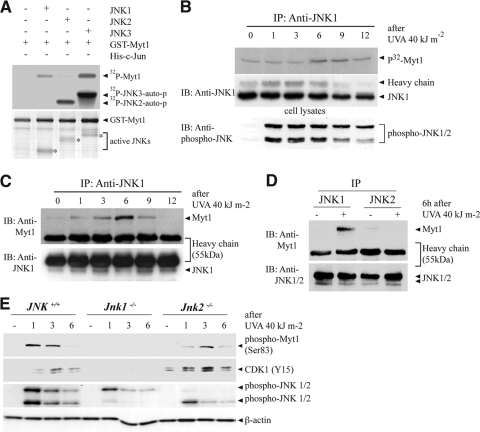
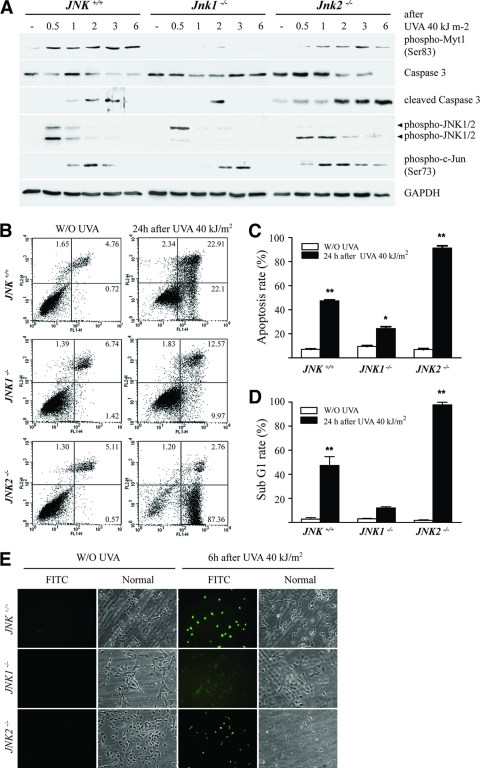
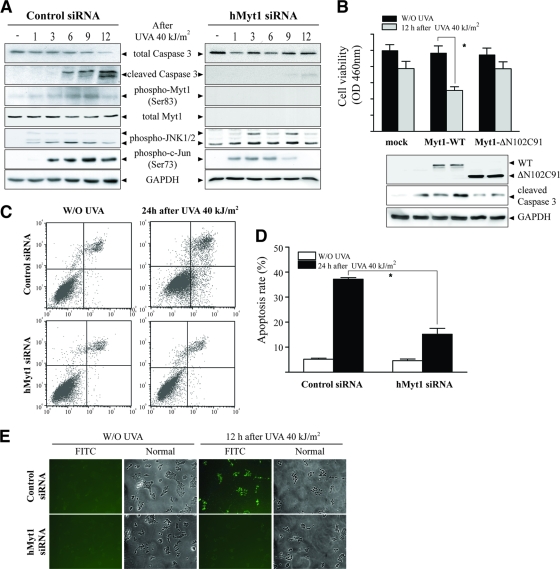
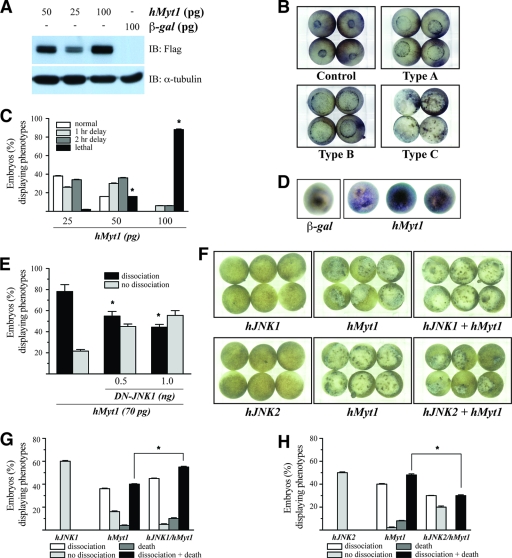
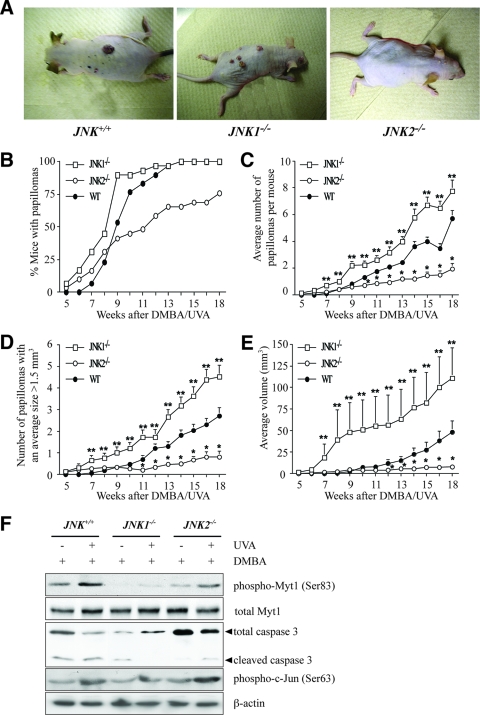
The c-Jun N-terminal kinase (JNK) signaling pathway is known to mediate both survival and apoptosis of tumor cells. Although JNK1 and JNK2 have been shown to differentially regulate the development of skin cancer, the underlying mechanistic basis remains unclear. Here, we demonstrate that JNK1, but not JNK2, interacts with and phosphorylates Myt1 ex vivo and in vitro. UVA induces substantial apoptosis in JNK wild-type (JNK(+/+)) or JNK2-deficient (JNK2(-/-)) mouse embryonic fibroblasts but has no effect on JNK1-deficient (JNK1(-/-)) cells. In addition, UVA-induced caspase-3 cleavage and DNA fragmentation were suppressed by the knockdown of human Myt1 in skin cancer cells. JNK1 deficiency results in suppressed Myt1 phosphorylation and caspase-3 cleavage in skin exposed to UVA irradiation. In contrast, the absence of JNK2 induces Myt1 phosphorylation and caspase-3 cleavage in skin exposed to UVA. The overexpression of JNK1 with Myt1 promotes cellular apoptosis during the early embryonic development of Xenopus laevis, whereas the presence of JNK2 reduces the phenotype of Myt1-induced apoptotic cell death. Most importantly, JNK1(-/-) mice developed more UVA-induced papillomas than either JNK(+/+) or JNK2(-/-) mice, which was associated with suppressed Myt1 phosphorylation and decreased caspase-3 cleavage. Taken together, these data provide mechanistic insights into the distinct roles of the different JNK isoforms, specifically suggesting that the JNK1-mediated phosphorylation of Myt1 plays an important role in UVA-induced apoptosis and the prevention of skin carcinogenesis.
Figures
References
-
- Bachelor, M. A., and G. T. Bowden. 2004. UVA-mediated activation of signaling pathways involved in skin tumor promotion and progression. Semin. Cancer Biol. 14131-138. - PubMed
-
- Caffrey, D. R., L. A. O'Neill, and D. C. Shields. 1999. The evolution of the MAP kinase pathways: coduplication of interacting proteins leads to new signaling cascades. J. Mol. Evol. 49567-582. - PubMed
-
- Carter, A. D., and J. C. Sible. 2003. Loss of XChk1 function triggers apoptosis after the midblastula transition in Xenopus laevis embryos. Mech. Dev. 120315-323. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous