

Estimation of genetic effects and genotype-phenotype maps
- PMID: 19204820
- PMCID: PMC2614198
- DOI: 10.4137/ebo.s756
Estimation of genetic effects and genotype-phenotype maps
Abstract
Determining the genetic architecture of complex traits is a necessary step to understand phenotypic changes in natural, experimental and domestic populations. However, this is still a major challenge for modern genetics, since the estimation of genetic effects tends to be complicated by genetic interactions, which lead to changes in the effect of allelic substitutions depending on the genetic background. Recent progress in statistical tools aiming to describe and quantify genetic effects meaningfully improves the efficiency and the availability of genotype-to-phenotype mapping methods. In this contribution, we facilitate the practical use of the recently published 'NOIA' quantitative framework by providing an implementation of linear and multilinear regressions, change of reference operation and genotype-to-phenotype mapping in a package ('noia') for the software R, and we discuss theoretical and practical benefits evolutionary and quantitative geneticists may find in using proper modeling strategies to quantify the effects of genes.
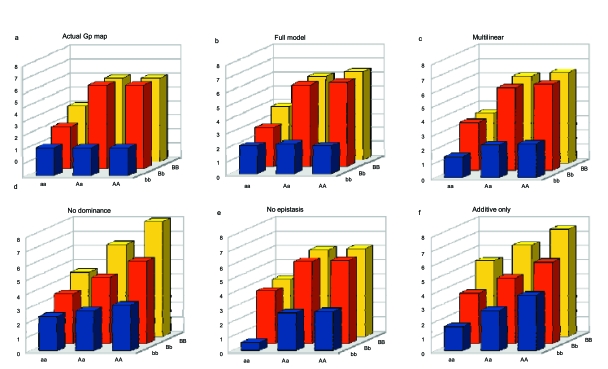
Figures
Similar articles
-
How to perform meaningful estimates of genetic effects.PLoS Genet. 2008 May 2;4(5):e1000062. doi: 10.1371/journal.pgen.1000062. PLoS Genet. 2008. PMID: 18451979 Free PMC article.
-
Is evolution predictable? Quantitative genetics under complex genotype-phenotype maps.Evolution. 2020 Feb;74(2):230-244. doi: 10.1111/evo.13907. Epub 2020 Jan 3. Evolution. 2020. PMID: 31883344
-
Mapping the genetic architecture of complex traits in experimental populations.Bioinformatics. 2007 Jun 15;23(12):1527-36. doi: 10.1093/bioinformatics/btm143. Epub 2007 Apr 25. Bioinformatics. 2007. PMID: 17459962
-
Estimation and interpretation of genetic effects with epistasis using the NOIA model.Methods Mol Biol. 2012;871:191-204. doi: 10.1007/978-1-61779-785-9_10. Methods Mol Biol. 2012. PMID: 22565838 Review.
-
Quantitative genetic methods depending on the nature of the phenotypic trait.Ann N Y Acad Sci. 2018 Jun;1422(1):29-47. doi: 10.1111/nyas.13571. Epub 2018 Jan 24. Ann N Y Acad Sci. 2018. PMID: 29363777 Review.
Cited by
-
Genetic architecture of tameness in a rat model of animal domestication.Genetics. 2009 Jun;182(2):541-54. doi: 10.1534/genetics.109.102186. Epub 2009 Apr 10. Genetics. 2009. PMID: 19363126 Free PMC article.
-
Dissection of the genetic architecture of body weight in chicken reveals the impact of epistasis on domestication traits.Genetics. 2008 Jul;179(3):1591-9. doi: 10.1534/genetics.108.089300. Epub 2008 Jul 13. Genetics. 2008. PMID: 18622035 Free PMC article.
-
Monotonicity is a key feature of genotype-phenotype maps.Front Genet. 2013 Nov 7;4:216. doi: 10.3389/fgene.2013.00216. eCollection 2013. Front Genet. 2013. PMID: 24223579 Free PMC article.
-
Mapping QTLs with main and epistatic effects underlying grain yield and heading time in soft winter wheat.Theor Appl Genet. 2011 Jul;123(2):283-92. doi: 10.1007/s00122-011-1583-y. Epub 2011 Apr 8. Theor Appl Genet. 2011. PMID: 21476040
-
Directionality of epistasis in a murine intercross population.Genetics. 2010 Aug;185(4):1489-505. doi: 10.1534/genetics.110.118356. Epub 2010 Jun 1. Genetics. 2010. PMID: 20516493 Free PMC article.
References
-
- Carter AJ, Hermisson J, Hansen TF. The role of epistatic gene interactions in the response to selection and the evolution of evolvability. Theor. Popul. Biol. 2005;68:179–96. - PubMed
LinkOut - more resources
Full Text Sources
