

Functional interactions between the ciliopathy-associated Meckel syndrome 1 (MKS1) protein and two novel MKS1-related (MKSR) proteins
- PMID: 19208769
- PMCID: PMC2720918
- DOI: 10.1242/jcs.028621
Functional interactions between the ciliopathy-associated Meckel syndrome 1 (MKS1) protein and two novel MKS1-related (MKSR) proteins
Abstract
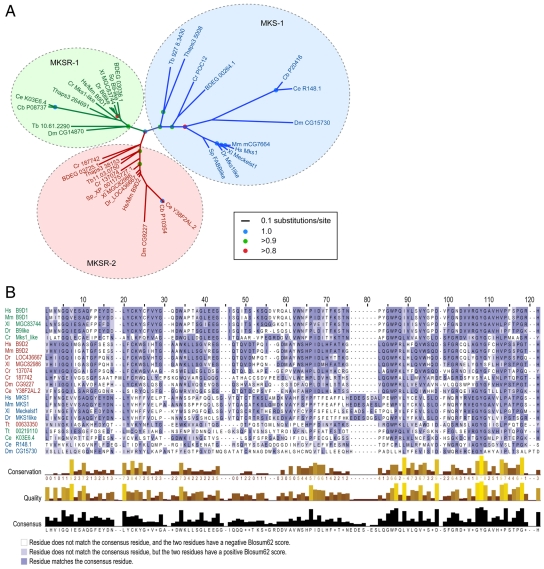
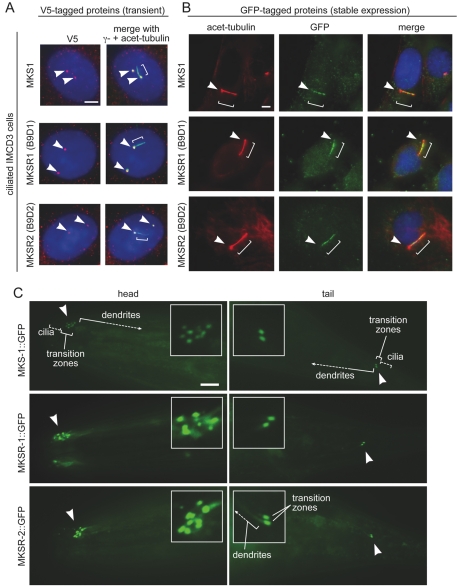
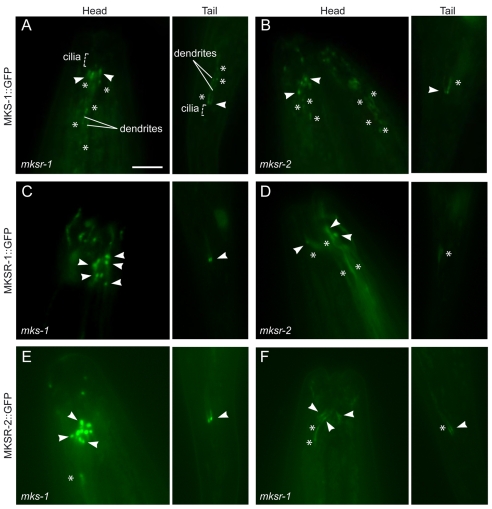
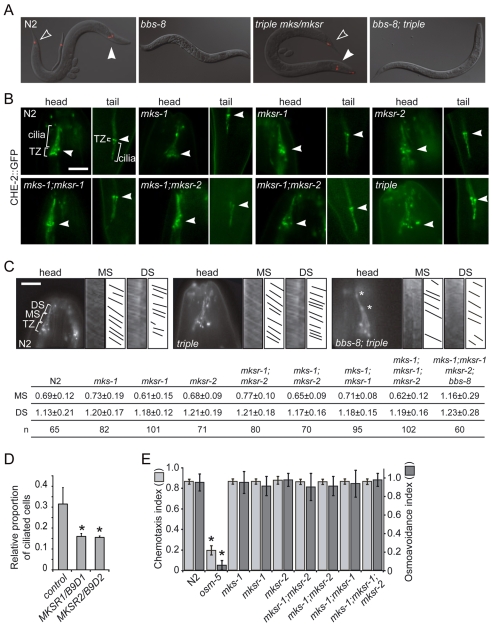
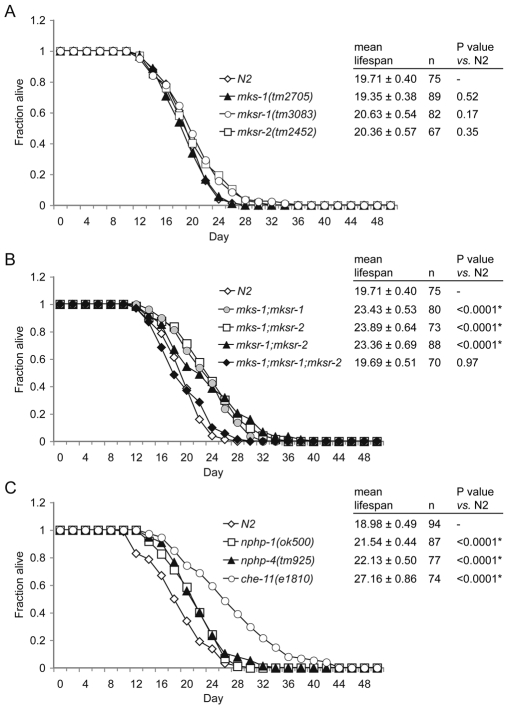
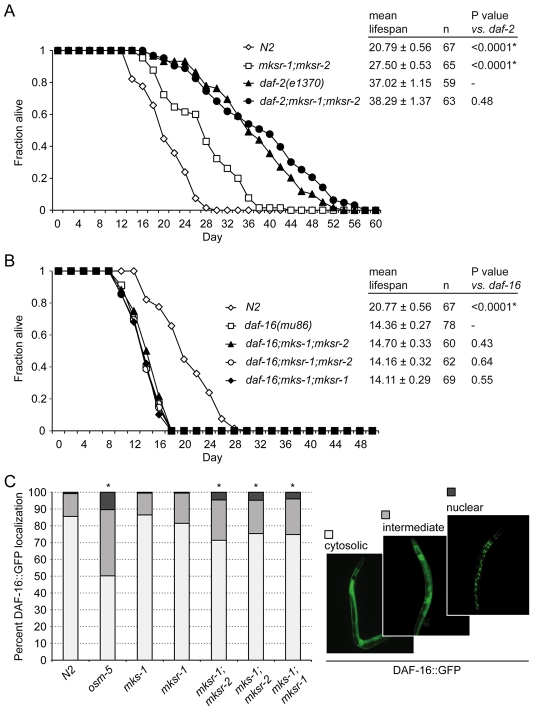
Meckel syndrome (MKS) is a ciliopathy characterized by encephalocele, cystic renal disease, liver fibrosis and polydactyly. An identifying feature of MKS1, one of six MKS-associated proteins, is the presence of a B9 domain of unknown function. Using phylogenetic analyses, we show that this domain occurs exclusively within a family of three proteins distributed widely in ciliated organisms. Consistent with a ciliary role, all Caenorhabditis elegans B9-domain-containing proteins, MKS-1 and MKS-1-related proteins 1 and 2 (MKSR-1, MKSR-2), localize to transition zones/basal bodies of sensory cilia. Their subcellular localization is largely co-dependent, pointing to a functional relationship between the proteins. This localization is evolutionarily conserved, because the human orthologues also localize to basal bodies, as well as cilia. As reported for MKS1, disrupting human MKSR1 or MKSR2 causes ciliogenesis defects. By contrast, single, double and triple C. elegans mks/mksr mutants do not display overt defects in ciliary structure, intraflagellar transport or chemosensation. However, we find genetic interactions between all double mks/mksr mutant combinations, manifesting as an increased lifespan phenotype, which is due to abnormal insulin-IGF-I signaling. Our findings therefore demonstrate functional interactions between a novel family of proteins associated with basal bodies or cilia, providing new insights into the molecular etiology of a pleiotropic human disorder.
Figures
References
-
- Alexiev, B. A., Lin, X., Sun, C. C. and Brenner, D. S. (2006). Meckel-Gruber syndrome: pathologic manifestations, minimal diagnostic criteria, and differential diagnosis. Arch. Pathol. Lab. Med. 130, 1236-1238. - PubMed
-
- Ansley, S. J., Badano, J. L., Blacque, O. E., Hill, J., Hoskins, B. E., Leitch, C. C., Kim, J. C., Ross, A. J., Eichers, E. R., Teslovich, T. M. et al. (2003). Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 425, 628-633. - PubMed
-
- Apfeld, J. and Kenyon, C. (1999). Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature 402, 804-809. - PubMed
-
- Arts, H. H., Doherty, D., van Beersum, S. E., Parisi, M. A., Letteboer, S. J., Gorden, N. T., Peters, T. A., Marker, T., Voesenek, K., Kartono, A. et al. (2007). Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 39, 882-888. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
