

Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing
- PMID: 19208812
- PMCID: PMC2638735
- DOI: 10.1073/pnas.0812841106
Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing
Abstract
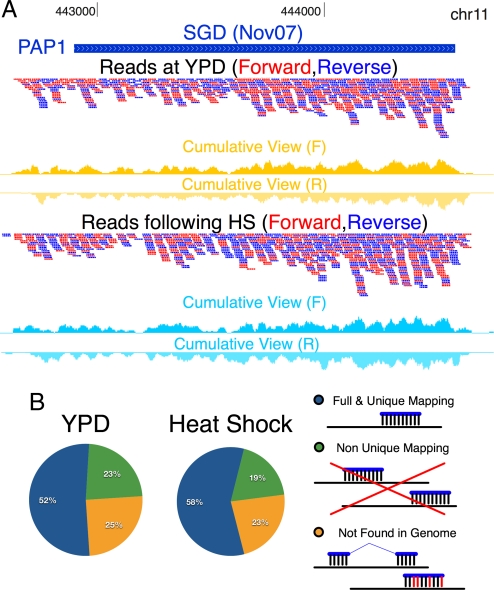
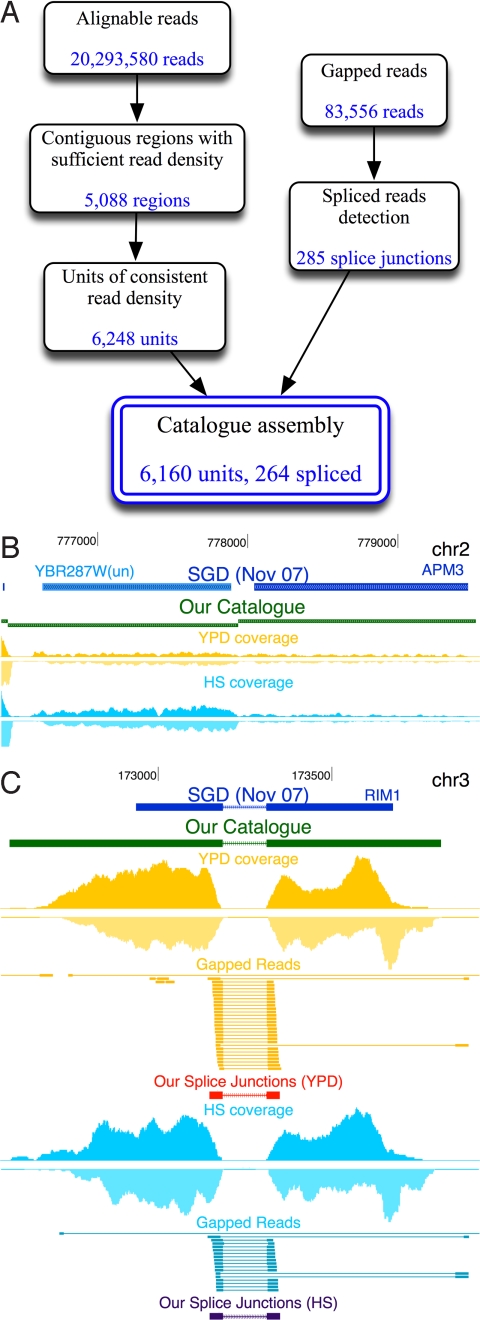
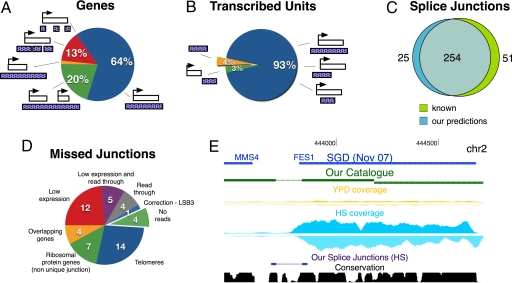
Defining the transcriptome, the repertoire of transcribed regions encoded in the genome, is a challenging experimental task. Current approaches, relying on sequencing of ESTs or cDNA libraries, are expensive and labor-intensive. Here, we present a general approach for ab initio discovery of the complete transcriptome of the budding yeast, based only on the unannotated genome sequence and millions of short reads from a single massively parallel sequencing run. Using novel algorithms, we automatically construct a highly accurate transcript catalog. Our approach automatically and fully defines 86% of the genes expressed under the given conditions, and discovers 160 previously undescribed transcription units of 250 bp or longer. It correctly demarcates the 5' and 3' UTR boundaries of 86 and 77% of expressed genes, respectively. The method further identifies 83% of known splice junctions in expressed genes, and discovers 25 previously uncharacterized introns, including 2 cases of condition-dependent intron retention. Our framework is applicable to poorly understood organisms, and can lead to greater understanding of the transcribed elements in an explored genome.
Conflict of interest statement
Conflict of interest statement: G.S., S.L., and I.K. are from Illumina Corporation, which developed the sequencing technology we used, and thus have competing financial interests.
Figures
References
-
- Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006;16:545–552. - PubMed
-
- Wilhelm BT, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–1243. - PubMed
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
