

Adenovirus induction of IRF3 occurs through a binary trigger targeting Jun N-terminal kinase and TBK1 kinase cascades and type I interferon autocrine signaling
- PMID: 19211767
- PMCID: PMC2668477
- DOI: 10.1128/JVI.02591-08
Adenovirus induction of IRF3 occurs through a binary trigger targeting Jun N-terminal kinase and TBK1 kinase cascades and type I interferon autocrine signaling
Abstract
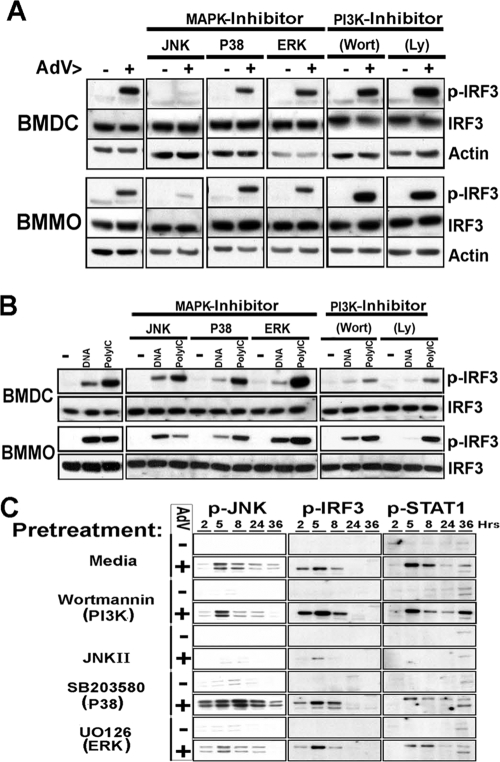
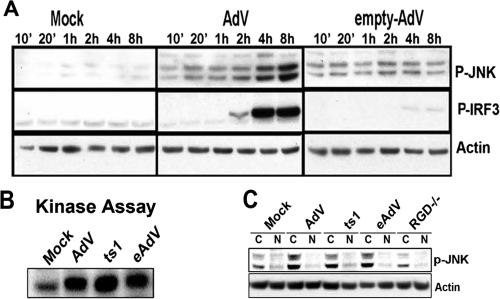
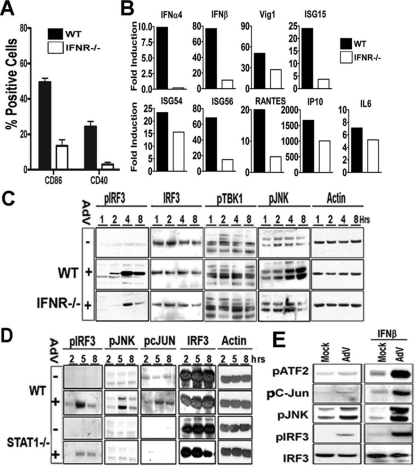
Pathogen recognition is a critical function of immune sentinel cells. Naïve macrophages or dendritic cells (DCs) undergo pathogen-directed activation and maturation, and as mature antigen-presenting cells (APCs), they contribute essential functions to both innate and adaptive immunity. Using recombinant adenovirus (rAdV) as a model for murine APC activation by DNA viruses, we demonstrate a critical role for stress kinase activation in cell intrinsic and extrinsic antiviral signaling cascades. We propose two viral triggers, viral capsid and viral DNA, are required for APC activation. Endosomal escape and presentation of cytosolic rAdV DNA induces phosphorylation of TANK-binding kinase 1 (TBK1) at serine 172 but does not induce IkappaB kinase epsilon activity as determined by in vitro kinase assays. However, induction of TBK1 alone is not sufficient for interferon regulatory factor 3 (IRF3) phosphorylation. We show that capsid-dependent activation of Jun N-terminal kinase (JNK) stress kinase is a necessary step, licensing TBK1 phosphorylation of IRF3 at Ser 396. A second later phase of JNK activity is required to coordinate phosphorylation of JNK-dependent transcription factors (c-Jun/ATF2) with activated IRF3 in the induction of primary IRF3-responsive transcripts. Finally, we demonstrate that maximal JNK/TBK1/IRF3 stimulation by rAdV depends on an intact type I interferon (IFN) signaling cascade. By requiring multiple viral triggers and type I IFN autocrine regulation, APCs have an inherent fail-safe mechanism against inappropriate activation and maturation.
Figures




References
-
- Agalioti, T., S. Lomvardas, B. Parekh, J. Yie, T. Maniatis, and D. Thanos. 2000. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell 103667-678. - PubMed
-
- Basner-Tschakarjan, E., E. Gaffal, M. O'Keeffe, D. Tormo, A. Limmer, H. Wagner, H. Hochrein, and T. Tuting. 2006. Adenovirus efficiently transduces plasmacytoid dendritic cells resulting in TLR9-dependent maturation and IFN-alpha production. J. Gene Med. 81300-1306. - PubMed
-
- Chen, W. C., and L. Huang. 2005. Non-viral vector as vaccine carrier. Adv. Genet. 54315-337. - PubMed
-
- Chu, W., X. Gong, Z. Li, K. Takabayashi, H. Ouyang, Y. Chen, A. Lois, D. J. Chen, G. C. Li, M. Karin, and E. Raz. 2000. DNA-PKcs is required for activation of innate immunity by immunostimulatory DNA. Cell 103909-918. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
