

The integrin antagonist cilengitide activates alphaVbeta3, disrupts VE-cadherin localization at cell junctions and enhances permeability in endothelial cells
- PMID: 19212436
- PMCID: PMC2636874
- DOI: 10.1371/journal.pone.0004449
The integrin antagonist cilengitide activates alphaVbeta3, disrupts VE-cadherin localization at cell junctions and enhances permeability in endothelial cells
Abstract
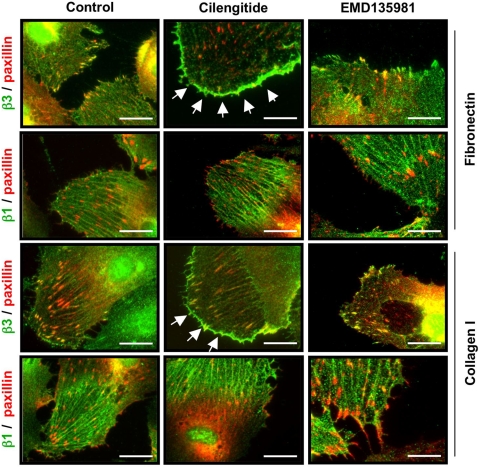
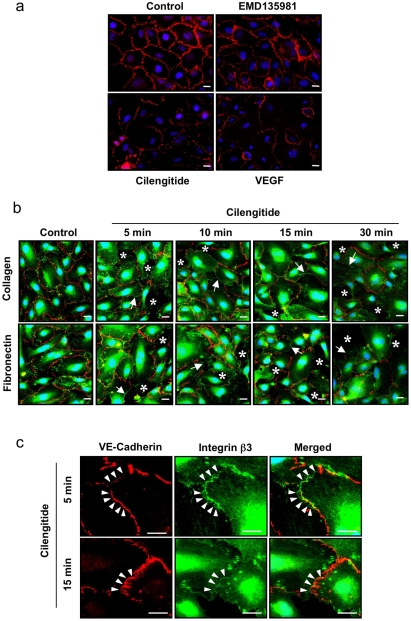
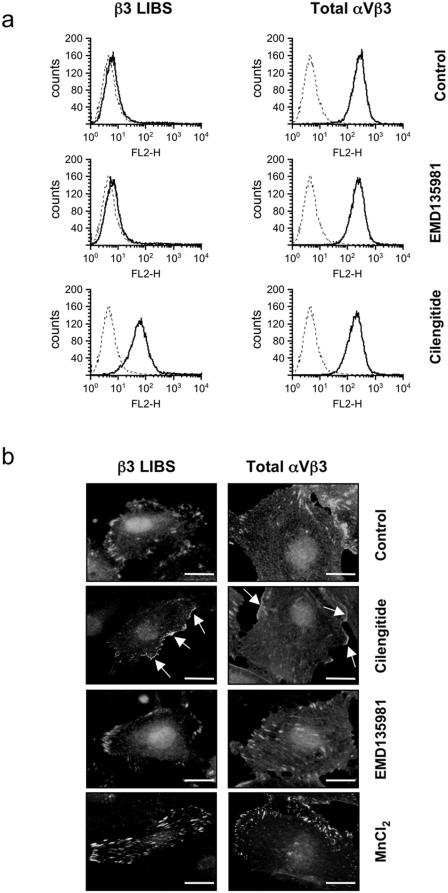
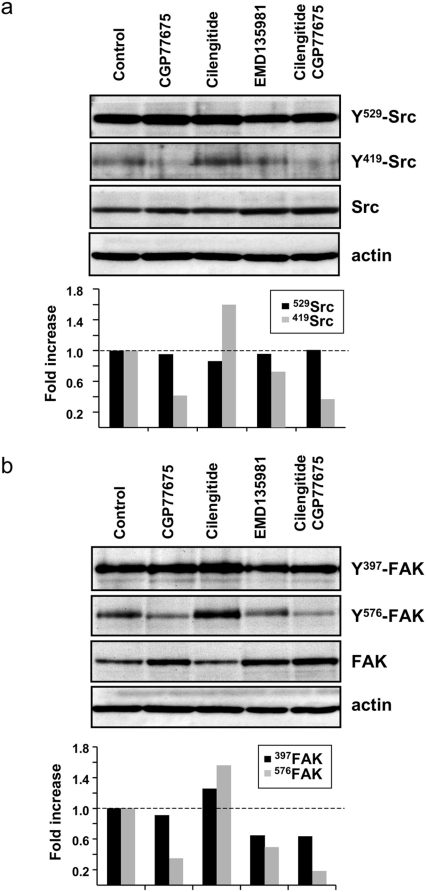
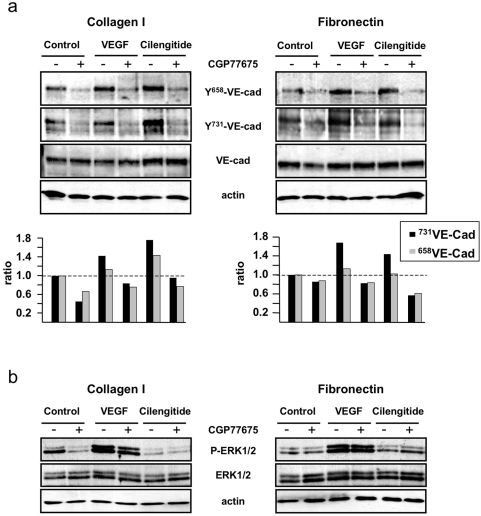
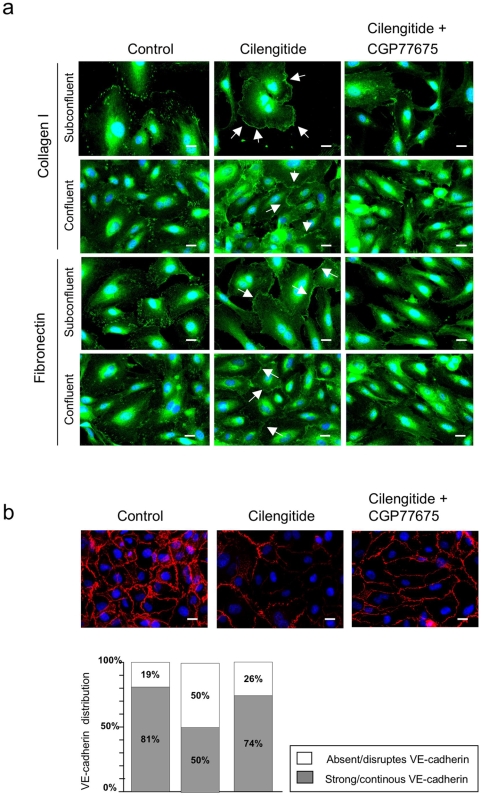
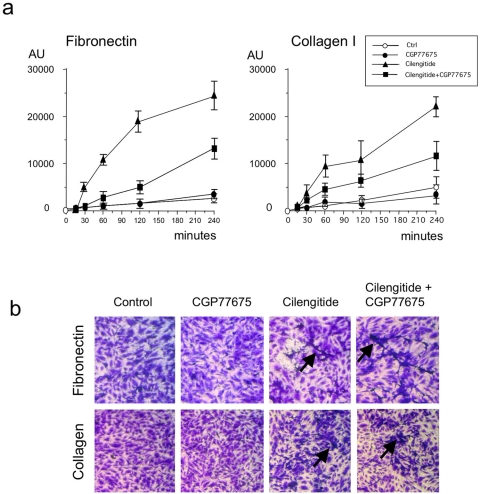
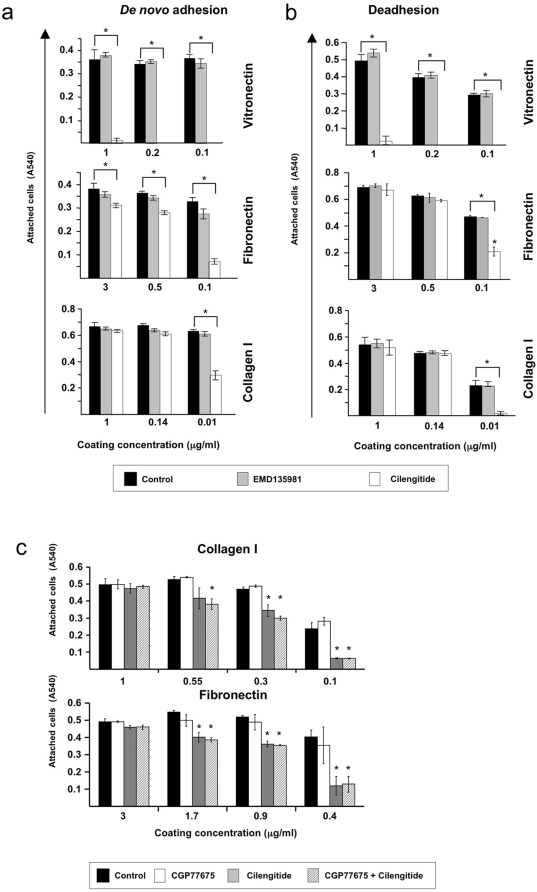
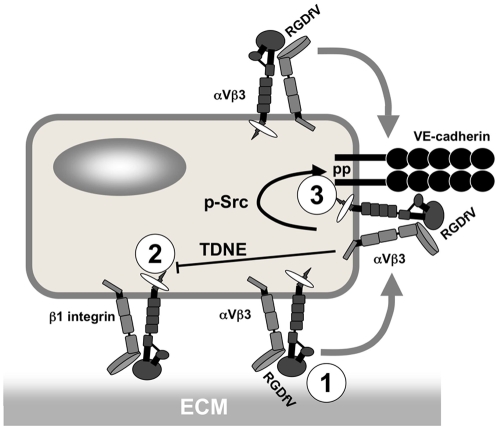
Cilengitide is a high-affinity cyclic pentapeptdic alphaV integrin antagonist previously reported to suppress angiogenesis by inducing anoikis of endothelial cells adhering through alphaVbeta3/alphaVbeta5 integrins. Angiogenic endothelial cells express multiple integrins, in particular those of the beta1 family, and little is known on the effect of cilengitide on endothelial cells expressing alphaVbeta3 but adhering through beta1 integrins. Through morphological, biochemical, pharmacological and functional approaches we investigated the effect of cilengitide on alphaVbeta3-expressing human umbilical vein endothelial cells (HUVEC) cultured on the beta1 ligands fibronectin and collagen I. We show that cilengitide activated cell surface alphaVbeta3, stimulated phosphorylation of FAK (Y(397) and Y(576/577)), Src (S(418)) and VE-cadherin (Y(658) and Y(731)), redistributed alphaVbeta3 at the cell periphery, caused disappearance of VE-cadherin from cellular junctions, increased the permeability of HUVEC monolayers and detached HUVEC adhering on low-density beta1 integrin ligands. Pharmacological inhibition of Src kinase activity fully prevented cilengitide-induced phosphorylation of Src, FAK and VE-cadherin, and redistribution of alphaVbeta3 and VE-cadherin and partially prevented increased permeability, but did not prevent HUVEC detachment from low-density matrices. Taken together, these observations reveal a previously unreported effect of cilengitide on endothelial cells namely its ability to elicit signaling events disrupting VE-cadherin localization at cellular contacts and to increase endothelial monolayer permeability. These effects are potentially relevant to the clinical use of cilengitide as anticancer agent.
Conflict of interest statement
Figures
References
-
- Hynes RO. Cell-matrix adhesion in vascular development. J Thromb Haemost. 2007;5(Suppl 1):32–40. - PubMed
-
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. - PubMed
-
- Alghisi GC, Rüegg C. Vascular Integrins in Tumor Angiogenesis: Mediators and Therapeutic Targets. Endothelium. 2006;13:113–135. - PubMed
-
- Kumar CC. Integrin alphavbeta3 as a Therapeutic Target for Blocking Tumor-Induced Angiogenesis. Current Drug Targets. 2003;4:123–131. - PubMed
-
- Smith J. Cilengitide Merck. Curr Opin Investig Drugs. 2003;4:741–745. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
