

Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants
- PMID: 19213730
- PMCID: PMC2665081
- DOI: 10.1074/jbc.M806907200
Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants
Abstract
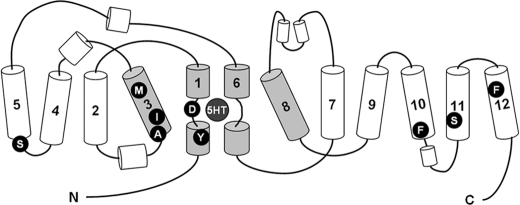
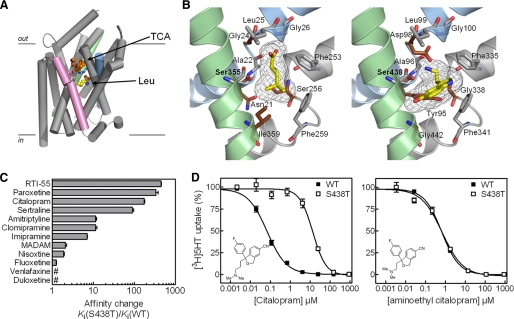
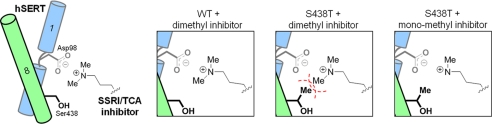
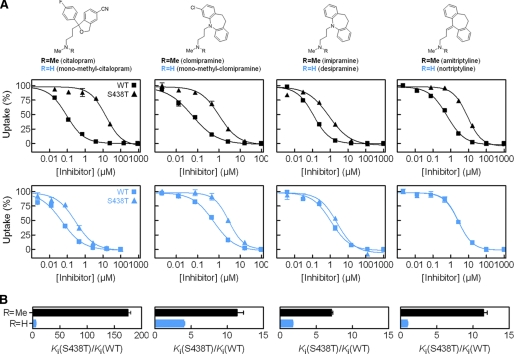
The serotonin transporter (SERT) regulates extracellular levels of serotonin (5-hydroxytryptamine, 5HT) in the brain by transporting 5HT into neurons and glial cells. The human SERT (hSERT) is the primary target for drugs used in the treatment of emotional disorders, including depression. hSERT belongs to the solute carrier 6 family that includes a bacterial leucine transporter (LeuT), for which a high resolution crystal structure has become available. LeuT has proved to be an excellent model for human transporters and has advanced the understanding of solute carrier 6 transporter structure-function relationships. However, the precise structural mechanism by which antidepressants inhibit hSERT and the location of their binding pockets are still elusive. We have identified a residue (Ser-438) located within the 5HT-binding pocket in hSERT to be a critical determinant for the potency of several antidepressants, including the selective serotonin reuptake inhibitor citalopram and the tricyclic antidepressants imipramine, clomipramine, and amitriptyline. A conservative mutation of Ser-438 to threonine (S438T) selectively increased the K(i) values for these antidepressants up to 175-fold. The effects of introducing a protein methyl group into the 5HT-binding pocket by S438T were absent or reduced for analogs of these antidepressants lacking a single methyl group. This suggests that these antidepressants interact directly with Ser-438 during binding to hSERT, implying an overlapping localization of substrate- and inhibitor-binding sites in hSERT suggesting that antidepressants function by a mechanism that involves direct occlusion of the 5HT-binding site.
Figures
References
-
- Wong, D. T., Perry, K. W., and Bymaster, F. P. (2005) Nat. Rev. Drug Discov. 4 764-774 - PubMed
-
- Yamashita, A., Singh, S. K., Kawate, T., Jin, Y., and Gouaux, E. (2005) Nature 437 215-223 - PubMed
-
- Vandenberg, R. J., Shaddick, K., and Ju, P. (2007) J. Biol. Chem. 282 14447-14453 - PubMed
-
- Dodd, J. R., and Christie, D. L. (2007) J. Biol. Chem. 282 15528-15533 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
