

Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo
- PMID: 19215222
- PMCID: PMC2677185
- DOI: 10.1042/BJ20081888
Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo
Abstract
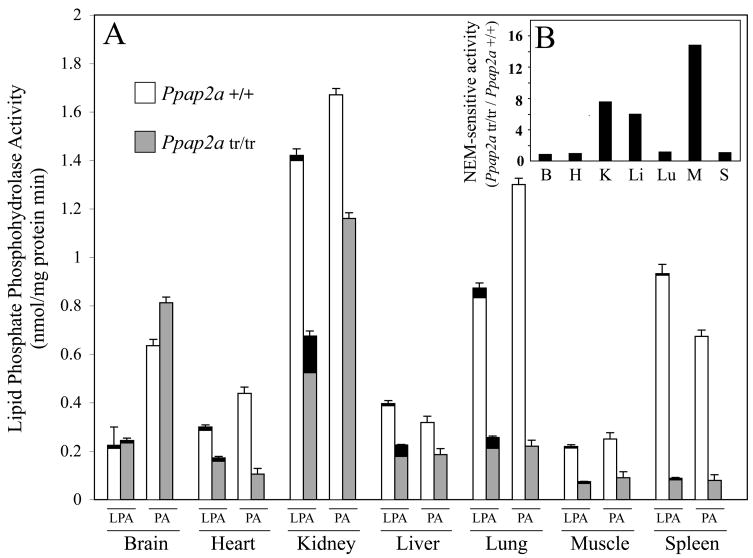
LPA (lysophosphatidic acid) is a lipid mediator that stimulates cell proliferation and growth, and is involved in physiological and pathological processes such as wound healing, platelet activation, angiogenesis and the growth of tumours. Therefore defining the mechanisms of LPA production and degradation are of interest in understanding the regulation of these processes. Extracellular LPA synthesis is relatively well understood, whereas the mechanisms of its degradation are not. One route of LPA degradation is dephosphorylation. A candidate enzyme is the integral membrane exophosphatase LPP1 (lipid phosphate phosphohydrolase type 1). In the present paper, we report the development of a mouse wherein the LPP1 gene (Ppap2a) was disrupted. The homozygous mice, which are phenotypically unremarkable, generally lack Ppap2a mRNA, and multiple tissues exhibit a substantial (35-95%) reduction in LPA phosphatase activity. Compared with wild-type littermates, Ppap2a(tr/tr) animals have increased levels of plasma LPA, and LPA injected intravenously is metabolized at a 4-fold lower rate. Our results demonstrate that LPA is rapidly metabolized in the bloodstream and that LPP1 is an important determinant of this turnover. These results indicate that LPP1 is a catabolic enzyme for LPA in vivo.
Figures






References
-
- Moolenaar WH. Development of our current understanding of bioactive lysophospholipids. Annals NY Acad Sci. 2000;905:1–10. - PubMed
-
- van Meeteren LA, Moolenaar WH. Regulation and biological activities of the autotaxin-LPA axis. Prog Lipid Res. 2007;46:145–160. - PubMed
-
- Anliker B, Chun J. Regulation and biological activities of the autotaxin-LPA axis. J Biol Chem. 2004;279:20555–20558. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous