

Genotype-imputation accuracy across worldwide human populations
- PMID: 19215730
- PMCID: PMC2668016
- DOI: 10.1016/j.ajhg.2009.01.013
Genotype-imputation accuracy across worldwide human populations
Abstract
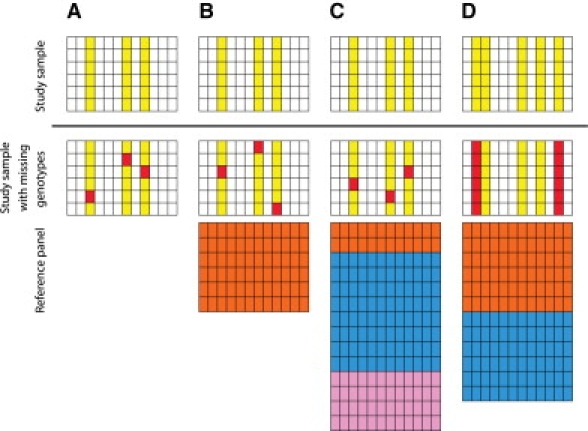
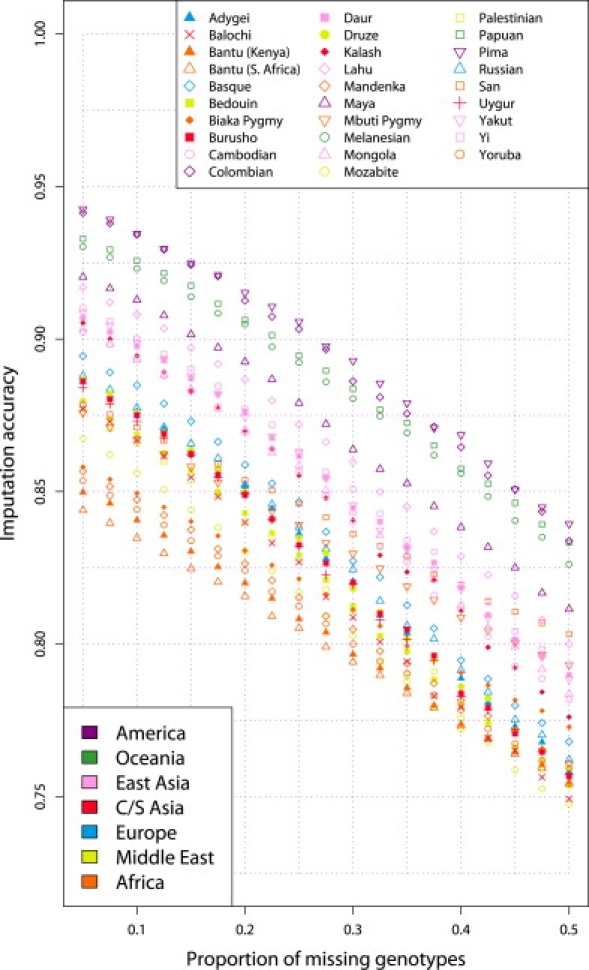
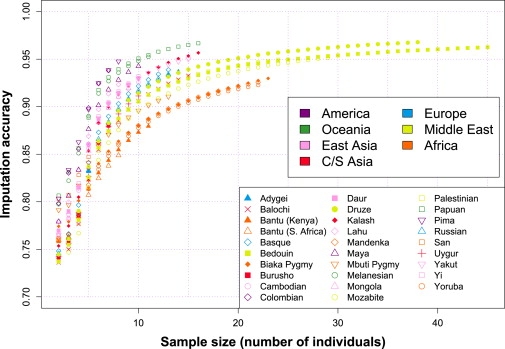
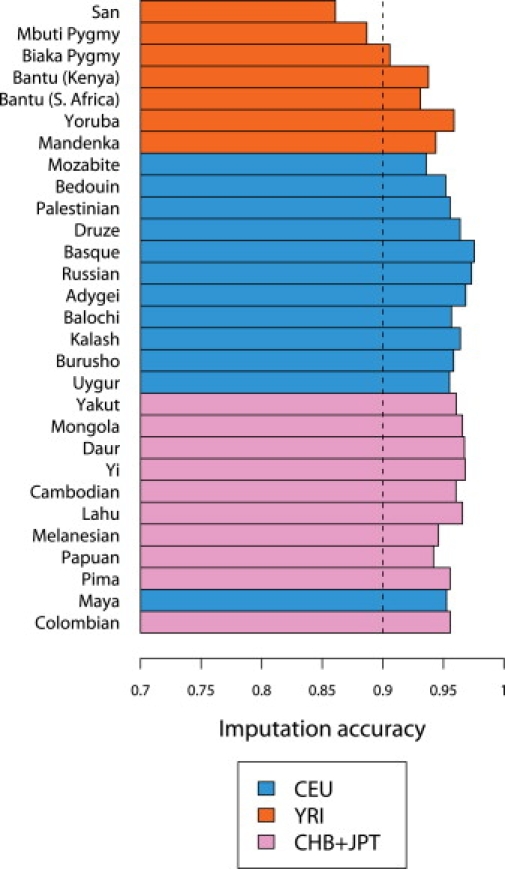
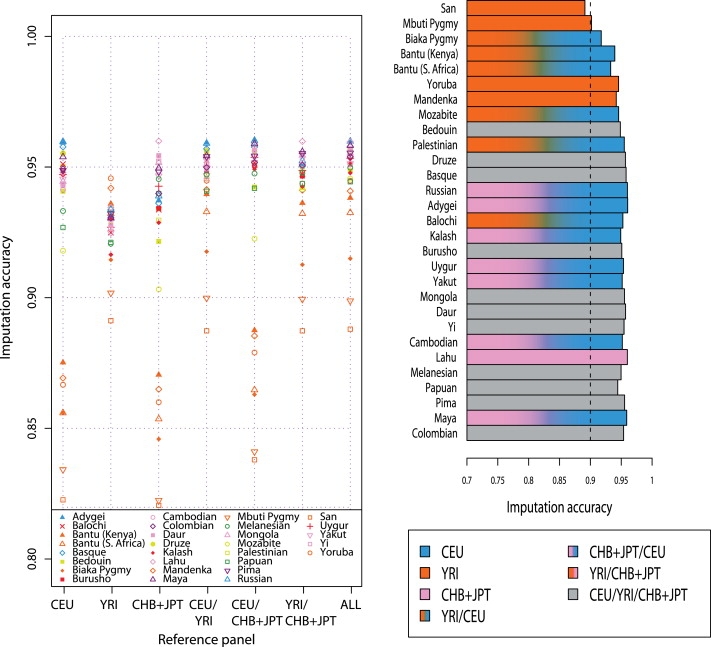
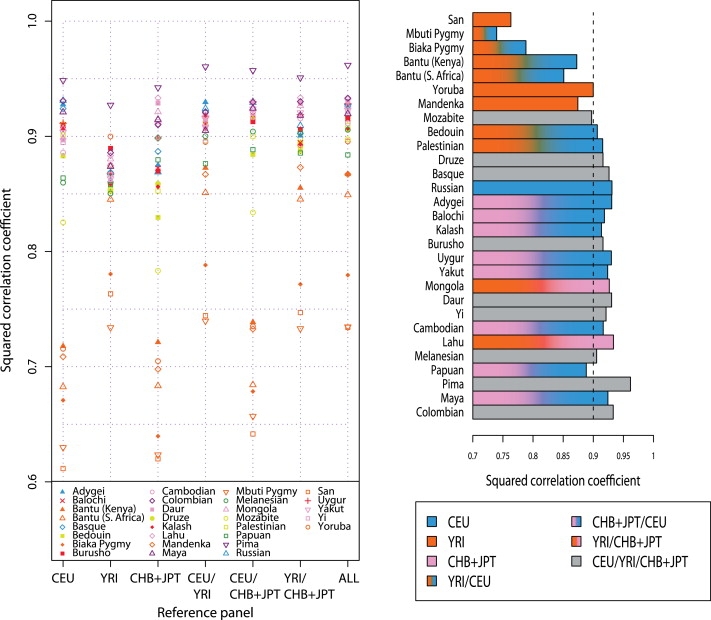
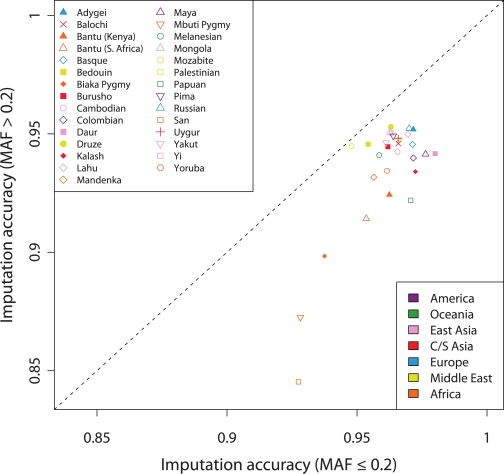
A current approach to mapping complex-disease-susceptibility loci in genome-wide association (GWA) studies involves leveraging the information in a reference database of dense genotype data. By modeling the patterns of linkage disequilibrium in a reference panel, genotypes not directly measured in the study samples can be imputed and tested for disease association. This imputation strategy has been successful for GWA studies in populations well represented by existing reference panels. We used genotypes at 513,008 autosomal single-nucleotide polymorphism (SNP) loci in 443 unrelated individuals from 29 worldwide populations to evaluate the "portability" of the HapMap reference panels for imputation in studies of diverse populations. When a single HapMap panel was leveraged for imputation of randomly masked genotypes, European populations had the highest imputation accuracy, followed by populations from East Asia, Central and South Asia, the Americas, Oceania, the Middle East, and Africa. For each population, we identified "optimal" mixtures of reference panels that maximized imputation accuracy, and we found that in most populations, mixtures including individuals from at least two HapMap panels produced the highest imputation accuracy. From a separate survey of additional SNPs typed in the same samples, we evaluated imputation accuracy in the scenario in which all genotypes at a given SNP position were unobserved and were imputed on the basis of data from a commercial "SNP chip," again finding that most populations benefited from the use of combinations of two or more HapMap reference panels. Our results can serve as a guide for selecting appropriate reference panels for imputation-based GWA analysis in diverse populations.
Figures


References
-
- Li Y., Ding J., Abecasis G.R. Mach 1.0: Rapid haplotype reconstruction and missing genotype inference. Am. J. Hum. Genet. 2006;79:S2290.
-
- Nicolae D.L. Testing untyped alleles (TUNA) - applications to genome-wide association studies. Genet. Epidemiol. 2006;30:718–727. - PubMed
-
- Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
