

Oncolytic adenoviral mutants with E1B19K gene deletions enhance gemcitabine-induced apoptosis in pancreatic carcinoma cells and anti-tumor efficacy in vivo
- PMID: 19223497
- PMCID: PMC2873675
- DOI: 10.1158/1078-0432.CCR-08-2008
Oncolytic adenoviral mutants with E1B19K gene deletions enhance gemcitabine-induced apoptosis in pancreatic carcinoma cells and anti-tumor efficacy in vivo
Abstract
Purpose: Pancreatic adenocarcinoma is a rapidly progressive malignancy that is highly resistant to current chemotherapeutic modalities and almost uniformly fatal. We show that a novel targeting strategy combining oncolytic adenoviral mutants with the standard cytotoxic treatment, gemcitabine, can markedly improve the anticancer potency.
Experimental design: Adenoviral mutants with the E1B19K gene deleted with and without E3B gene expression (AdDeltaE1B19K and dl337 mutants, respectively) were assessed for synergistic interactions in combination with gemcitabine. Cell viability, mechanism of cell death, and antitumor efficacy in vivo were determined in the pancreatic carcinoma cells PT45 and Suit2, normal human bronchial epithelial cells, and in PT45 xenografts.
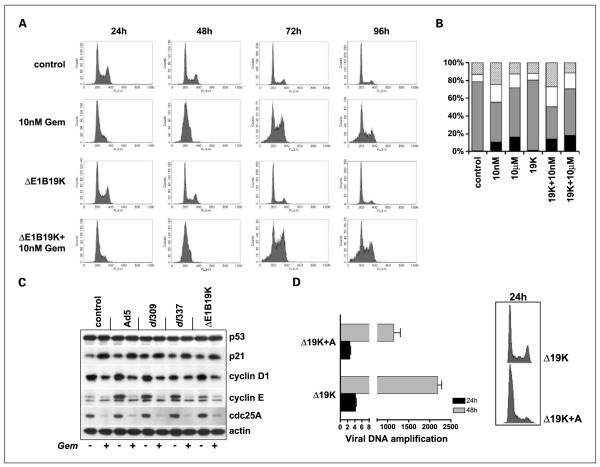
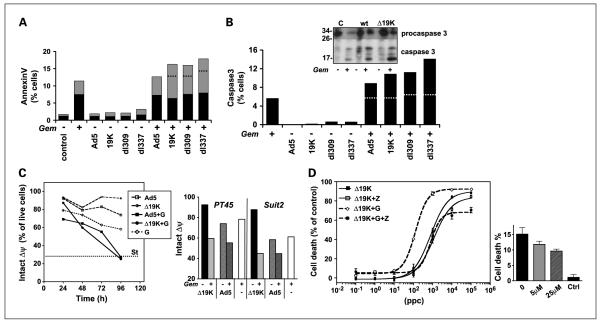
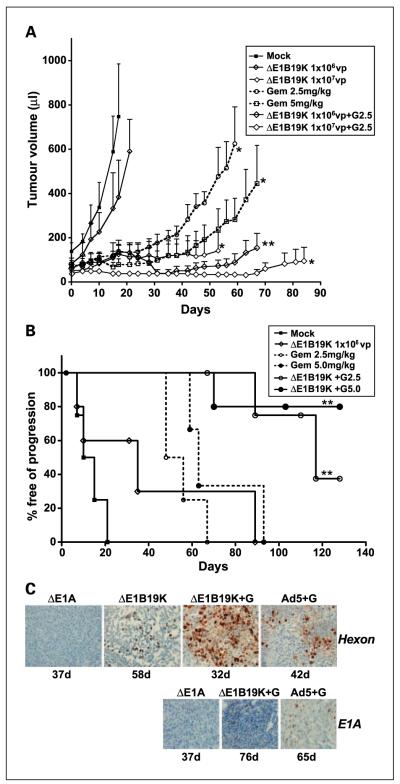
Results: The DeltaE1B19K-deleted mutants synergized with gemcitabine to selectively kill cultured pancreatic cancer cells and xenografts in vivo with no effect in normal cells. The corresponding wild-type virus (Ad5) stimulated drug-induced cell killing to a lesser degree. Gemcitabine blocked replication of all viruses despite the enhanced cell killing activity due to gemcitabine-induced delay in G1/S-cell cycle progression, with repression of cyclin E and cdc25A, which was not abrogated by viral E1A-expression. Synergistic cell death occurred through enhancement of gemcitabine-induced apoptosis in the presence of both AdDeltaE1B19K and dl337 mutants, shown by increased cell membrane fragmentation, caspase-3 activation, and mitochondrial dysfunction.
Conclusions: Our data suggest that oncolytic mutants lacking the antiapoptotic E1B19K gene can improve efficacy of DNA-damaging drugs such as gemcitabine through convergence on cellular apoptosis pathways. These findings imply that less toxic doses than currently practiced in the clinic could efficiently target pancreatic adenocarcinomas when combined with adenoviral mutants.
Figures
Similar articles
-
Improved potency and selectivity of an oncolytic E1ACR2 and E1B19K deleted adenoviral mutant in prostate and pancreatic cancers.Clin Cancer Res. 2010 Jan 15;16(2):541-53. doi: 10.1158/1078-0432.CCR-09-1960. Epub 2010 Jan 12. Clin Cancer Res. 2010. PMID: 20068104 Free PMC article.
-
Optimisation of replication-selective oncolytic adenoviral mutants in combination with chemotherapeutics.J BUON. 2009 Sep;14 Suppl 1:S61-7. J BUON. 2009. PMID: 19785071 Review.
-
The oncolytic adenovirus AdΔΔ enhances selective cancer cell killing in combination with DNA-damaging drugs in pancreatic cancer models.Gene Ther. 2011 Dec;18(12):1157-65. doi: 10.1038/gt.2011.141. Epub 2011 Oct 6. Gene Ther. 2011. PMID: 21975464
-
An oncolytic adenovirus defective in pRb-binding (dl922-947) can efficiently eliminate pancreatic cancer cells and tumors in vivo in combination with 5-FU or gemcitabine.Cancer Gene Ther. 2011 Oct;18(10):734-43. doi: 10.1038/cgt.2011.45. Epub 2011 Aug 12. Cancer Gene Ther. 2011. PMID: 21836633
-
Advances in oncolytic adenovirus therapy for pancreatic cancer.Cancer Lett. 2018 Oct 10;434:56-69. doi: 10.1016/j.canlet.2018.07.006. Epub 2018 Jul 5. Cancer Lett. 2018. PMID: 29981812 Review.
Cited by
-
Recent advances in oncolytic virus design.Clin Transl Oncol. 2011 Apr;13(4):229-39. doi: 10.1007/s12094-011-0647-4. Clin Transl Oncol. 2011. PMID: 21493183 Review.
-
Complexing the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T with Cationic Nanoparticles Enhances Viral Infection and Spread in Prostate and Pancreatic Cancer Models.Int J Mol Sci. 2022 Aug 10;23(16):8884. doi: 10.3390/ijms23168884. Int J Mol Sci. 2022. PMID: 36012152 Free PMC article.
-
Expanding the Spectrum of Pancreatic Cancers Responsive to Vesicular Stomatitis Virus-Based Oncolytic Virotherapy: Challenges and Solutions.Cancers (Basel). 2021 Mar 9;13(5):1171. doi: 10.3390/cancers13051171. Cancers (Basel). 2021. PMID: 33803211 Free PMC article. Review.
-
Microorganisms in chemotherapy for pancreatic cancer: An overview of current research and future directions.Int J Biol Sci. 2021 Jun 26;17(10):2666-2682. doi: 10.7150/ijbs.59117. eCollection 2021. Int J Biol Sci. 2021. PMID: 34326701 Free PMC article. Review.
-
Development of a novel oncolytic adenovirus controlled by CDX2 promoter for esophageal adenocarcinoma therapy.J Gastroenterol. 2024 Nov;59(11):986-999. doi: 10.1007/s00535-024-02147-2. Epub 2024 Sep 4. J Gastroenterol. 2024. PMID: 39227437
References
-
- Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. - PubMed
-
- Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–77. - PubMed
-
- Kasuya H, Takeda S, Nomoto S, Nakao A. The potential of oncolytic virus therapy for pancreatic cancer. Cancer GeneTher. 2005;12:725–36. - PubMed
-
- O’Shea CC, Johnson L, Bagus B, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611–23. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
