

Tumor suppressor protein C53 antagonizes checkpoint kinases to promote cyclin-dependent kinase 1 activation
- PMID: 19223857
- PMCID: PMC2664857
- DOI: 10.1038/cr.2009.14
Tumor suppressor protein C53 antagonizes checkpoint kinases to promote cyclin-dependent kinase 1 activation
Abstract
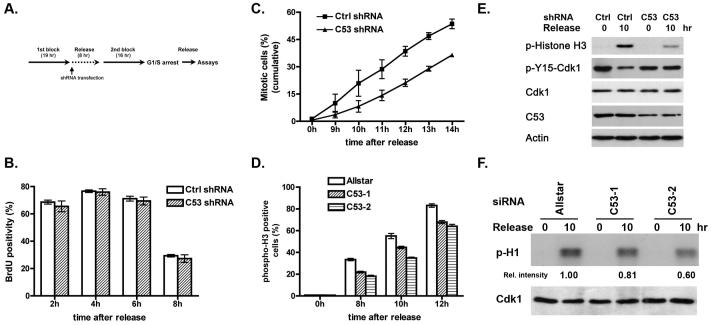
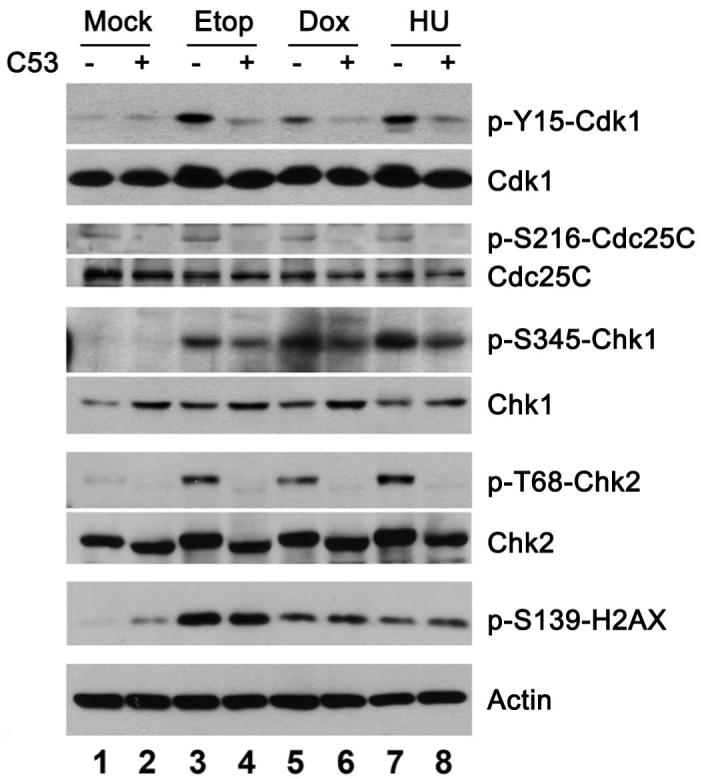
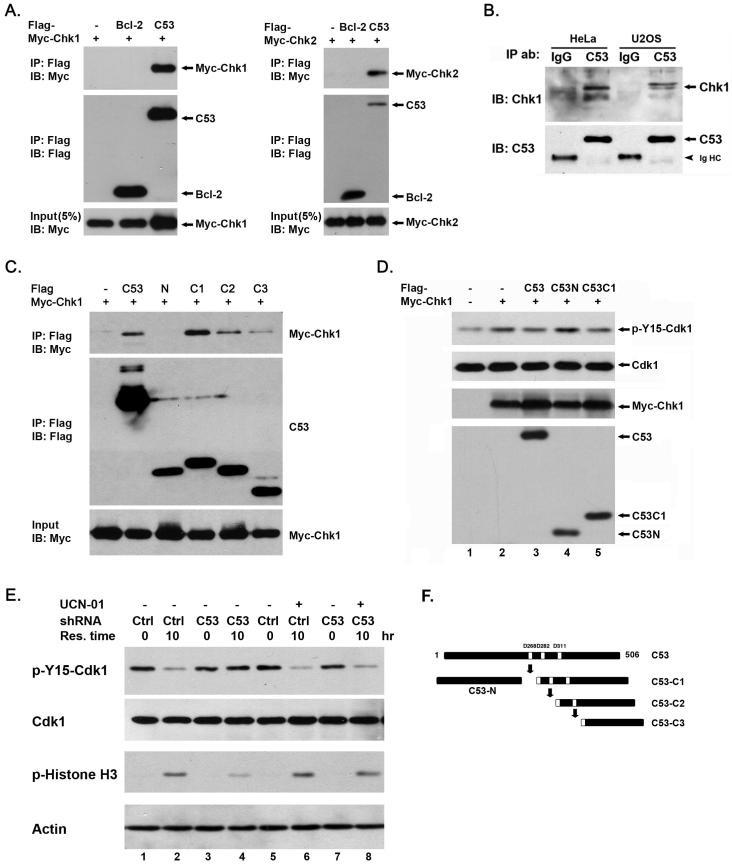
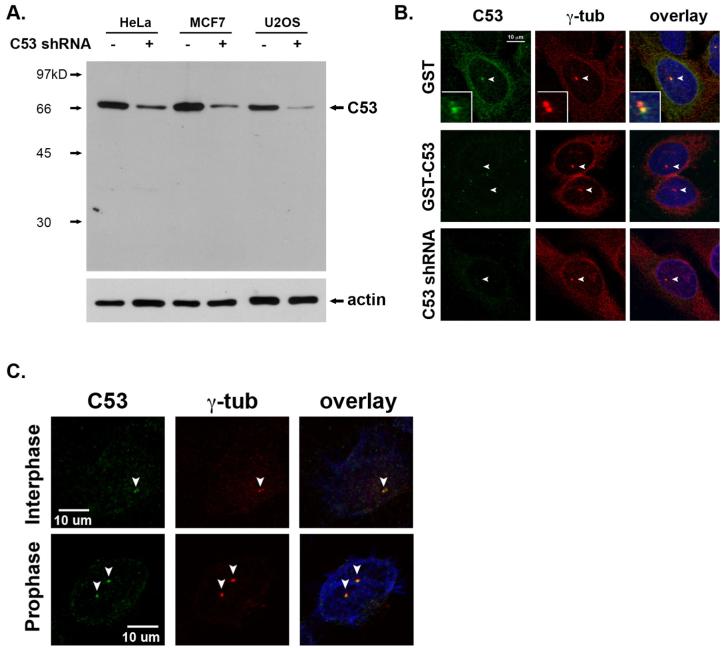
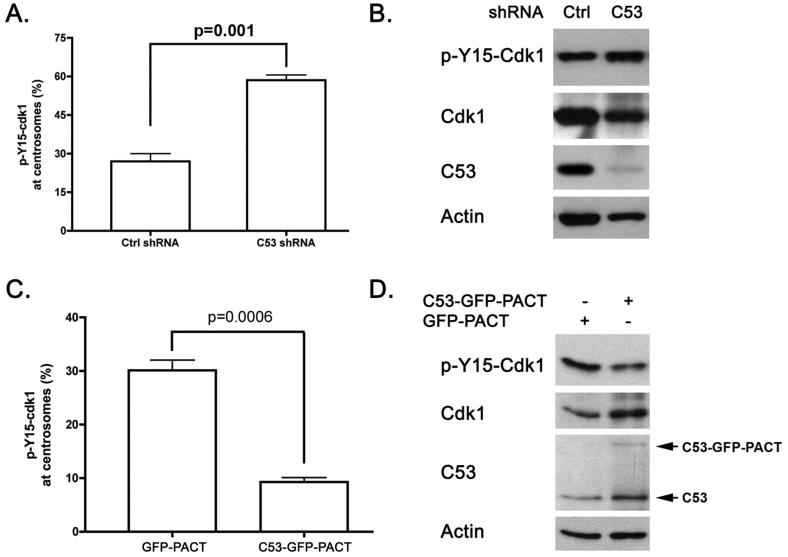
Cyclin-dependent kinase 1 (Cdk1)/cyclin B1 complex is the driving force for mitotic entry, and its activation is tightly regulated by the G2/M checkpoint. We originally reported that a novel protein C53 (also known as Cdk5rap3 and LZAP) potentiates DNA damage-induced cell death by modulating the G2/M checkpoint. More recently, Wang et al. (2007) found that C53/LZAP may function as a tumor suppressor by way of inhibiting NF-kappaB signaling. We report here the identification of C53 protein as a novel regulator of Cdk1 activation. We found that knockdown of C53 protein causes delayed Cdk1 activation and mitotic entry. During DNA damage response, activation of checkpoint kinase 1 and 2 (Chk1 and Chk2) is partially inhibited by C53 overexpression. Intriguingly, we found that C53 interacts with Chk1 and antagonizes its function. Moreover, a portion of C53 protein is localized at the centrosome, and centrosome-targeting C53 potently promotes local Cdk1 activation. Taken together, our results strongly suggest that C53 is a novel negative regulator of checkpoint response. By counteracting Chk1, C53 promotes Cdk1 activation and mitotic entry in both unperturbed cell-cycle progression and DNA damage response.
Figures
Similar articles
-
Cdk5 activator-binding protein C53 regulates apoptosis induced by genotoxic stress via modulating the G2/M DNA damage checkpoint.J Biol Chem. 2005 May 27;280(21):20651-9. doi: 10.1074/jbc.M413431200. Epub 2005 Mar 24. J Biol Chem. 2005. PMID: 15790566
-
Interaction of LHBs with C53 promotes hepatocyte mitotic entry: A novel mechanism for HBV-induced hepatocellular carcinoma.Oncol Rep. 2012 Jan;27(1):151-9. doi: 10.3892/or.2011.1489. Epub 2011 Oct 4. Oncol Rep. 2012. PMID: 21971960
-
The cell cycle checkpoint kinase Chk2 is a negative regulator of mitotic catastrophe.Oncogene. 2004 May 27;23(25):4353-61. doi: 10.1038/sj.onc.1207573. Oncogene. 2004. PMID: 15048074
-
Regulation of the G2/M transition by p53.Oncogene. 2001 Apr 5;20(15):1803-15. doi: 10.1038/sj.onc.1204252. Oncogene. 2001. PMID: 11313928 Review.
-
DNA damage: Chk1 and Cdc25, more than meets the eye.Curr Opin Genet Dev. 2001 Feb;11(1):78-82. doi: 10.1016/s0959-437x(00)00160-x. Curr Opin Genet Dev. 2001. PMID: 11163155 Review.
Cited by
-
The UFMylation System in Proteostasis and Beyond.Trends Cell Biol. 2019 Dec;29(12):974-986. doi: 10.1016/j.tcb.2019.09.005. Epub 2019 Nov 6. Trends Cell Biol. 2019. PMID: 31703843 Free PMC article. Review.
-
A novel LZAP-binding protein, NLBP, inhibits cell invasion.J Biol Chem. 2010 Apr 16;285(16):12232-40. doi: 10.1074/jbc.M109.065920. Epub 2010 Feb 17. J Biol Chem. 2010. PMID: 20164180 Free PMC article.
-
Network Topologies Decoding Cervical Cancer.PLoS One. 2015 Aug 26;10(8):e0135183. doi: 10.1371/journal.pone.0135183. eCollection 2015. PLoS One. 2015. PMID: 26308848 Free PMC article.
-
Tumor suppressor Lzap regulates cell cycle progression, doming, and zebrafish epiboly.Dev Dyn. 2011 Jun;240(6):1613-25. doi: 10.1002/dvdy.22644. Epub 2011 Apr 26. Dev Dyn. 2011. PMID: 21523853 Free PMC article.
-
CDK5RAP3, a New BRCA2 Partner That Regulates DNA Repair, Is Associated with Breast Cancer Survival.Cancers (Basel). 2022 Jan 12;14(2):353. doi: 10.3390/cancers14020353. Cancers (Basel). 2022. PMID: 35053516 Free PMC article.
References
-
- Jiang H, Luo S, Li H. Cdk5 Activator-binding Protein C53 Regulates Apoptosis Induced by Genotoxic Stress via Modulating the G2/M DNA Damage Checkpoint. J Biol Chem. 2005;280:20651–20659. - PubMed
-
- Wang J, An H, Mayo MW, Baldwin AS, Yarbrough WG. LZAP, a putative tumor suppressor, selectively inhibits NF-kappaB. Cancer Cell. 2007;12:239–251. - PubMed
-
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. - PubMed
-
- Shiloh Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet. 1997;31:635–662. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
