

Saposin B-dependent reconstitution of arylsulfatase A activity in vitro and in cell culture models of metachromatic leukodystrophy
- PMID: 19224915
- PMCID: PMC2666589
- DOI: 10.1074/jbc.M809457200
Saposin B-dependent reconstitution of arylsulfatase A activity in vitro and in cell culture models of metachromatic leukodystrophy
Abstract
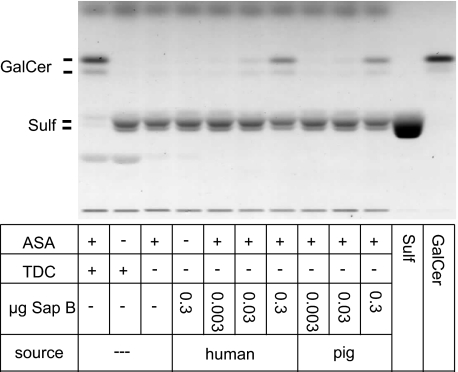
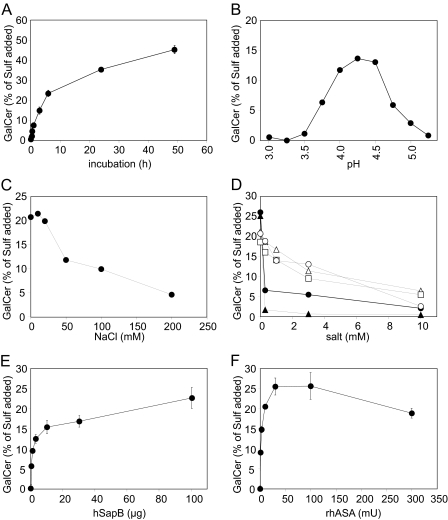
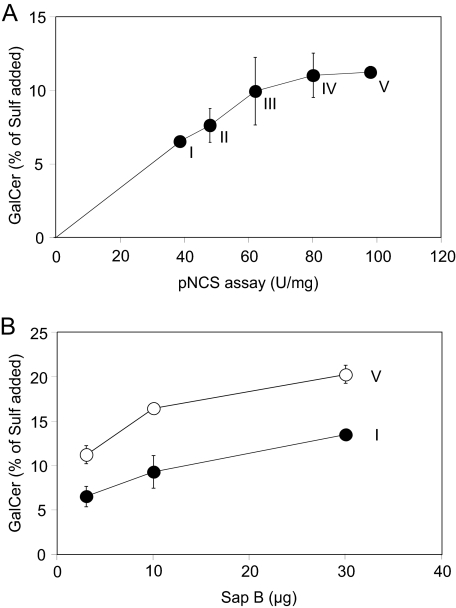
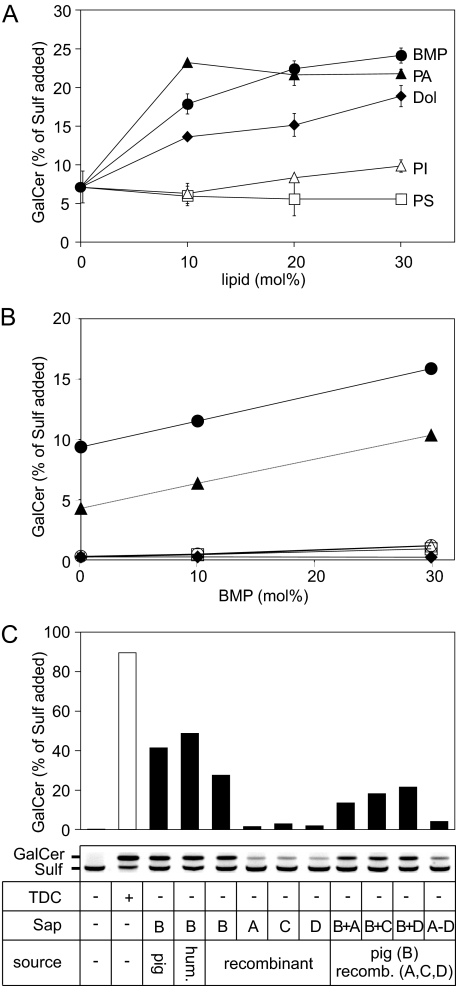
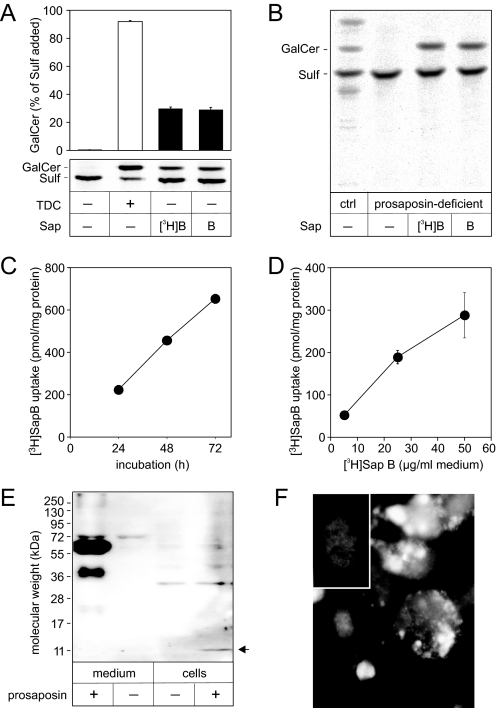
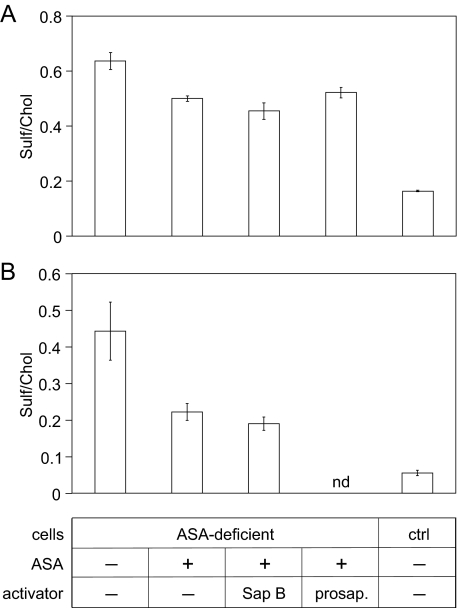
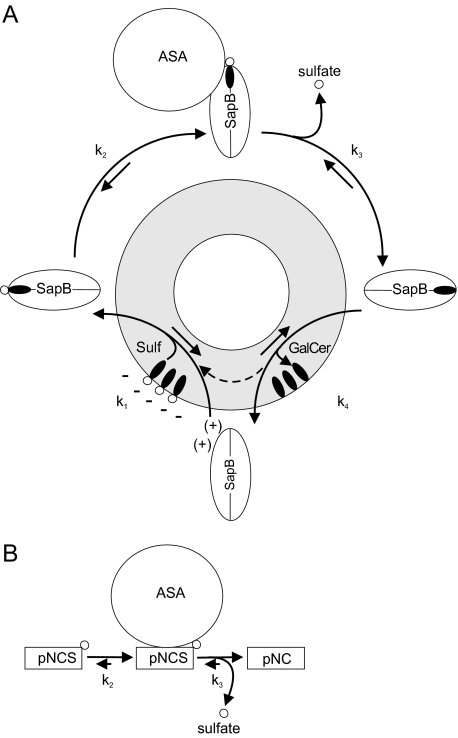
Arylsulfatase A (ASA) catalyzes the intralysosomal desulfation of 3-O-sulfogalactosylceramide (sulfatide) to galactosylceramide. The reaction requires saposin B (Sap B), a non-enzymatic proteinaceous cofactor which presents sulfatide to the catalytic site of ASA. The lack of either ASA or Sap B results in a block of sulfatide degradation, progressive intralysosomal accumulation of sulfatide, and the fatal lysosomal storage disease metachromatic leukodystrophy. We studied the coupled Sap B-ASA reaction in vitro using detergent-free micellar and liposomal assay systems and in vivo using cell culture models of metachromatic leukodystrophy. Under in vitro conditions, the reaction had a narrow pH optimum around pH 4.3 and was inhibited by mono- and divalent cations, phosphate and sulfite. Bis(monoacylglycero) phosphate and phosphatidic acid were activators of the reaction, underscoring a significant role of acidic phosphoglycerolipids in sphingolipid degradation. Desulfation was negligible when Sap B was substituted by Sap A, C, or D. Up to a molar ratio between Sap B and sulfatide of 1:5, an elevation of Sap B concentrations caused a sharp increase of sulfatide hydrolysis, indicating the requirement of unexpected high Sap B levels for maximum turnover. Feeding of ASA-deficient, sulfatide-storing primary mouse kidney cells with ASA caused partial clearance of sulfatide. Co-feeding of Sap B or its precursor prosaposin resulted in the lysosomal uptake of the cofactor but did not promote ASA-catalyzed sulfatide hydrolysis. This suggests that Sap B is not a limiting factor of the coupled Sap B-ASA reaction in mouse kidney cells even if sulfatide has accumulated to unphysiologically high levels.
Figures
References
-
- Kolter, T., and Sandhoff, K. (2005) Annu. Rev. Cell Dev. Biol. 21 81-103 - PubMed
-
- Sandhoff, K., Kolter, T., and Harzer, K. (2001) in The Metabolic and Molecular Basis of Inherited Disease (Scriver, C. R., Beaudet, A. L., Sly, W. S., and Valle, D., eds) pp. 3371-3388, McGraw-Hill, New York
-
- Kishimoto, Y., Hiraiwa, M., and O'Brien, J. S. (1992) J. Lipid Res. 33 1255-1267 - PubMed
-
- Fujita, N., Suzuki, K., Vanier, M. T., Popko, B., Maeda, N., Klein, A., Henseler, M., Sandhoff, K., Nakayasu, H., and Suzuki, K. (1996) Hum. Mol. Genet. 5 711-725 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
