

Cellular mediators of renal vascular dysfunction in hypertension
- PMID: 19225145
- PMCID: PMC2698613
- DOI: 10.1152/ajpregu.90960.2008
Cellular mediators of renal vascular dysfunction in hypertension
Abstract
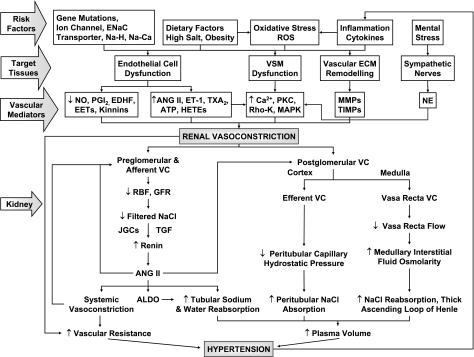
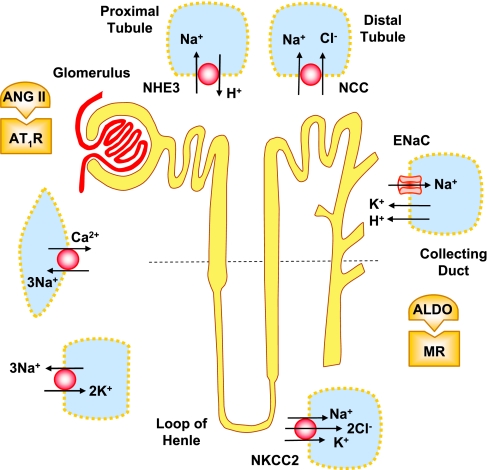
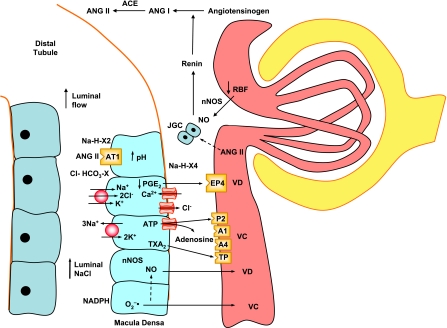
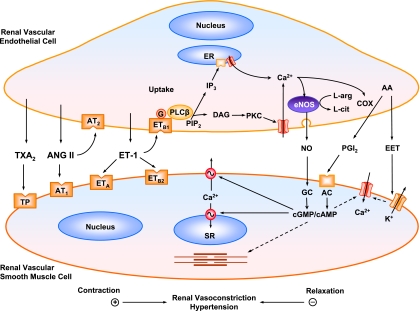
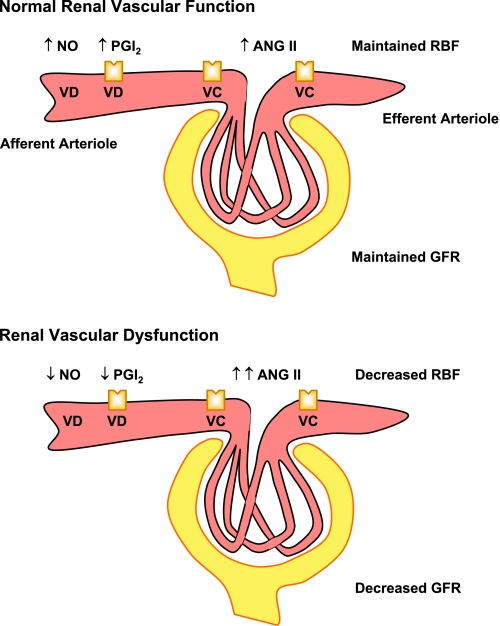
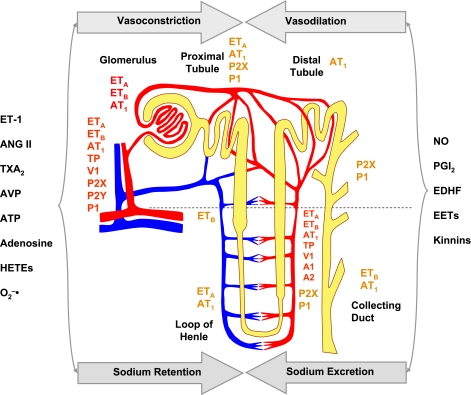
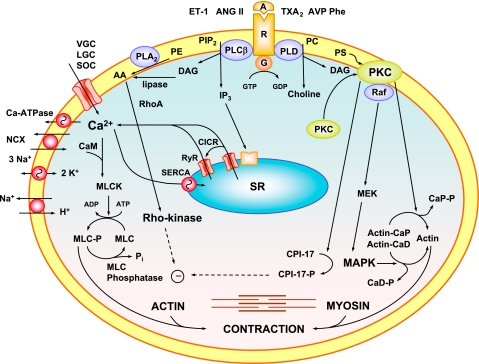
The renal vasculature plays a major role in the regulation of renal blood flow and the ability of the kidney to control the plasma volume and blood pressure. Renal vascular dysfunction is associated with renal vasoconstriction, decreased renal blood flow, and consequent increase in plasma volume and has been demonstrated in several forms of hypertension (HTN), including genetic and salt-sensitive HTN. Several predisposing factors and cellular mediators have been implicated, but the relationship between their actions on the renal vasculature and the consequent effects on renal tubular function in the setting of HTN is not clearly defined. Gene mutations/defects in an ion channel, a membrane ion transporter, and/or a regulatory enzyme in the nephron and renal vasculature may be a primary cause of renal vascular dysfunction. Environmental risk factors, such as high dietary salt intake, vascular inflammation, and oxidative stress further promote renal vascular dysfunction. Renal endothelial cell dysfunction is manifested as a decrease in the release of vasodilatory mediators, such as nitric oxide, prostacyclin, and hyperpolarizing factors, and/or an increase in vasoconstrictive mediators, such as endothelin, angiotensin II, and thromboxane A(2). Also, an increase in the amount/activity of intracellular Ca(2+) concentration, protein kinase C, Rho kinase, and mitogen-activated protein kinase in vascular smooth muscle promotes renal vasoconstriction. Matrix metalloproteinases and their inhibitors could also modify the composition of the extracellular matrix and lead to renal vascular remodeling. Synergistic interactions between the genetic and environmental risk factors on the cellular mediators of renal vascular dysfunction cause persistent renal vasoconstriction, increased renal vascular resistance, and decreased renal blood flow, and, consequently, lead to a disturbance in the renal control mechanisms of water and electrolyte balance, increased plasma volume, and HTN. Targeting the underlying genetic defects, environmental risk factors, and the aberrant renal vascular mediators involved should provide complementary strategies in the management of HTN.
Figures
References
-
- Agarwal A, Williams GH, Fisher ND. Genetics of human hypertension. Trends Endocrinol Metab 16: 127–133, 2005. - PubMed
-
- Aldred KL, Harris PJ, Eitle E. Increased proximal tubule NHE-3 and H+-ATPase activities in spontaneously hypertensive rats. J Hypertens 18: 623–628, 2000. - PubMed
-
- Alonso-Galicia M, Maier KG, Greene AS, Cowley AW Jr, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid in the renal and vasoconstrictor actions of angiotensin II. Am J Physiol Regul Integr Comp Physiol 283: R60–R68, 2002. - PubMed
-
- An SJ, Boyd R, Zhu M, Chapman A, Pimentel DR, Wang HD. NADPH oxidase mediates angiotensin II-induced endothelin-1 expression in vascular adventitial fibroblasts. Cardiovasc Res 75:702–709, 2007. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous