

Differential susceptibility to excitotoxic stress in YAC128 mouse models of Huntington disease between initiation and progression of disease
- PMID: 19228972
- PMCID: PMC2729178
- DOI: 10.1523/JNEUROSCI.5473-08.2009
Differential susceptibility to excitotoxic stress in YAC128 mouse models of Huntington disease between initiation and progression of disease
Abstract
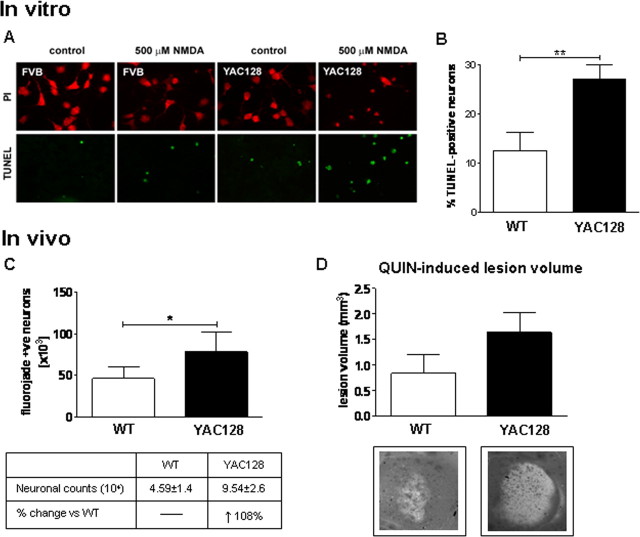
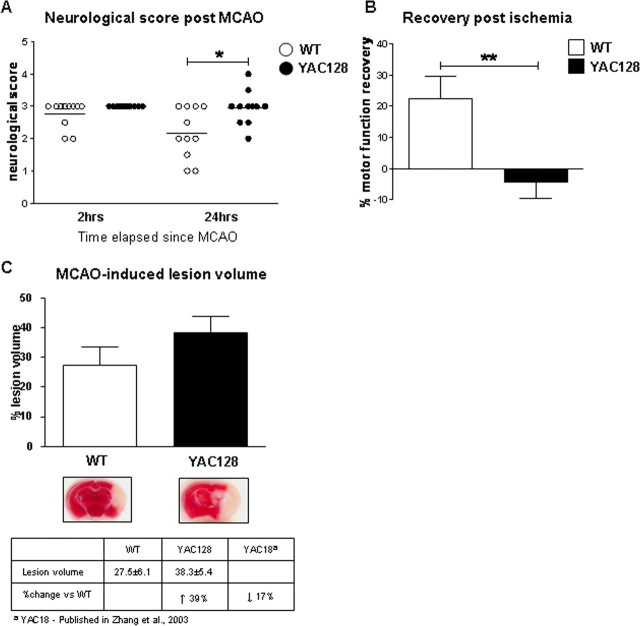
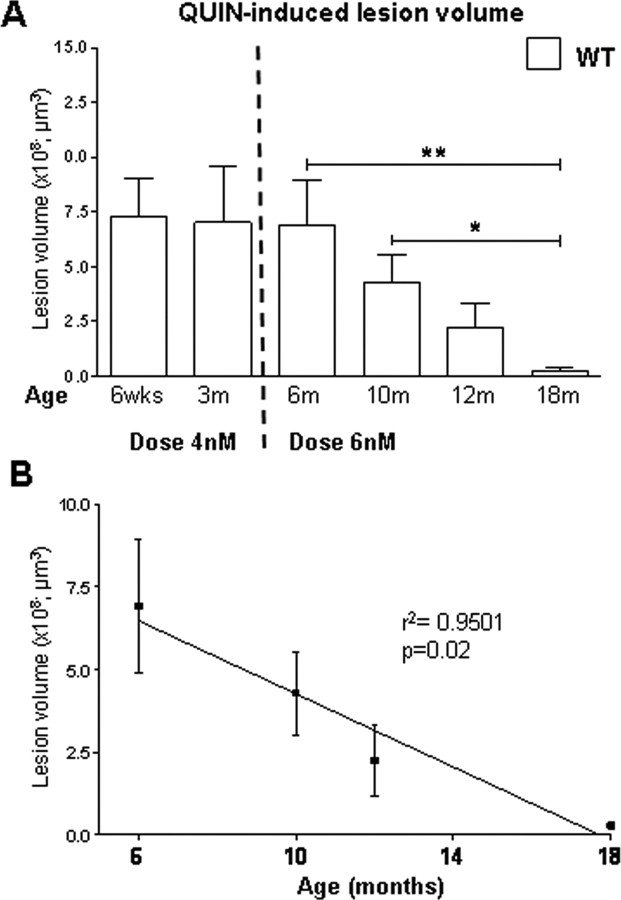
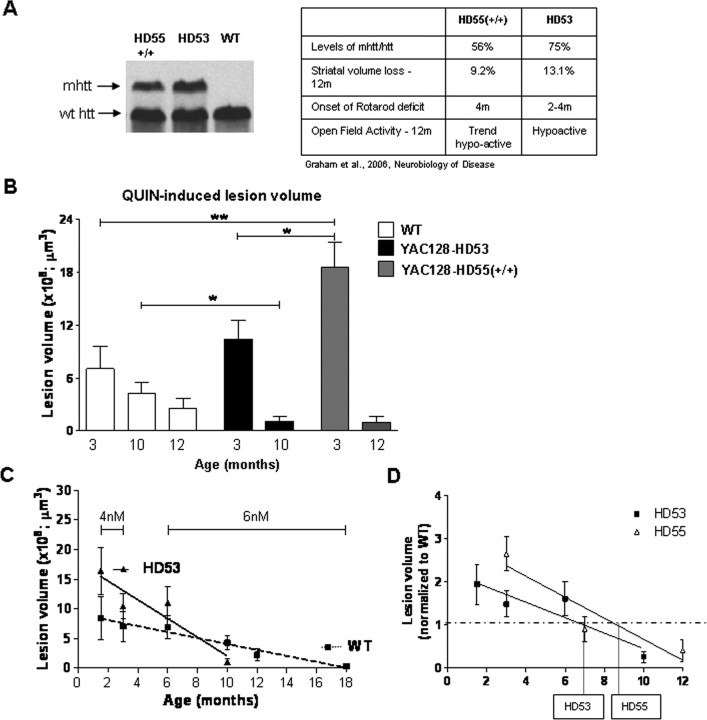
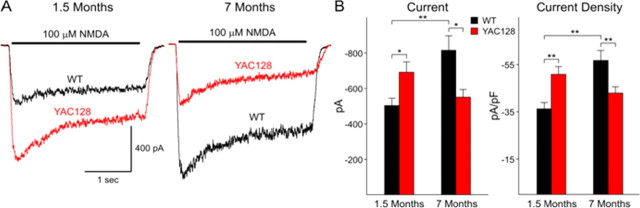
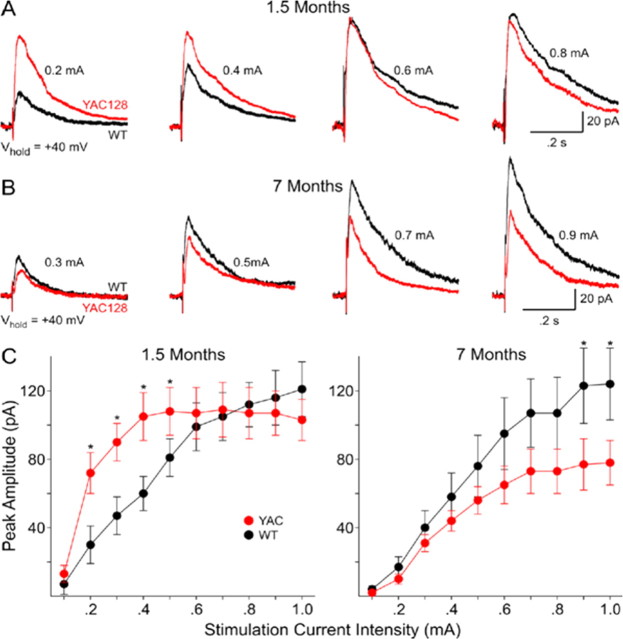
Huntington disease (HD) is a neurodegenerative disorder caused by an expanded CAG tract in the HD gene. Polyglutamine expansion of huntingtin (htt) results in early, progressive loss of medium spiny striatal neurons, as well as cortical neurons that project to the striatum. Excitotoxicity has been postulated to play a key role in the selective vulnerability of striatal neurons in HD. Early excitotoxic neuropathological changes observed in human HD brain include increased quinolinate (QUIN) concurrent with proliferative changes such as increased spine density and dendritic length. In later stages of the disease, degenerative-type changes are apparent, such as loss of dendritic arborization, a reduction in spine density and reduced levels of 3-hydroxykynurenine and QUIN. It is currently unknown whether sensitivity to excitotoxic stress varies between initiation and progression of disease. Here, we have assessed the excitotoxic phenotype in the YAC128 mouse model of HD by examining the response to excitotoxic stress at different stages of disease. Our results demonstrate that YAC128 mice display enhanced sensitivity to NMDA ex vivo and QUIN in vivo before obvious phenotypic changes. In contrast, 10-month-old symptomatic YAC128 mice are resistant to QUIN-induced neurotoxicity. These findings are paralleled by a significant increase in NMDAR-mediated membrane currents in presymptomatic YAC128 dissociated medium spiny neurons progressing to reduced NMDAR-mediated membrane currents with disease progression. These data highlight the dynamic nature of the mutant htt-mediated excitotoxic phenotype and suggests that therapeutic approaches to HD may need to be altered, depending on the stage and development of the disease.
Figures
Similar articles
-
Phosphorylation of huntingtin at Ser421 in YAC128 neurons is associated with protection of YAC128 neurons from NMDA-mediated excitotoxicity and is modulated by PP1 and PP2A.J Neurosci. 2010 Oct 27;30(43):14318-29. doi: 10.1523/JNEUROSCI.1589-10.2010. J Neurosci. 2010. PMID: 20980587 Free PMC article.
-
Altering cortical input unmasks synaptic phenotypes in the YAC128 cortico-striatal co-culture model of Huntington disease.BMC Biol. 2018 Jun 27;16(1):58. doi: 10.1186/s12915-018-0526-3. BMC Biol. 2018. PMID: 29945611 Free PMC article.
-
Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington's disease.Neurobiol Dis. 2008 Jul;31(1):80-8. doi: 10.1016/j.nbd.2008.03.010. Epub 2008 Apr 16. Neurobiol Dis. 2008. PMID: 18502655 Free PMC article.
-
Selective degeneration in YAC mouse models of Huntington disease.Brain Res Bull. 2007 Apr 30;72(2-3):124-31. doi: 10.1016/j.brainresbull.2006.10.018. Epub 2006 Nov 16. Brain Res Bull. 2007. PMID: 17352936 Review.
-
Selective neuronal degeneration in Huntington's disease.Curr Top Dev Biol. 2006;75:25-71. doi: 10.1016/S0070-2153(06)75002-5. Curr Top Dev Biol. 2006. PMID: 16984809 Review.
Cited by
-
Onset and progression of behavioral and molecular phenotypes in a novel congenic R6/2 line exhibiting intergenerational CAG repeat stability.PLoS One. 2011;6(12):e28409. doi: 10.1371/journal.pone.0028409. Epub 2011 Dec 7. PLoS One. 2011. PMID: 22163300 Free PMC article.
-
C57BL/6 Background Attenuates mHTT Toxicity in the Striatum of YAC128 Mice.Int J Mol Sci. 2021 Nov 23;22(23):12664. doi: 10.3390/ijms222312664. Int J Mol Sci. 2021. PMID: 34884469 Free PMC article.
-
Effects of excitotoxicity in the hypothalamus in transgenic mouse models of Huntington disease.Heliyon. 2021 Aug 14;7(8):e07808. doi: 10.1016/j.heliyon.2021.e07808. eCollection 2021 Aug. Heliyon. 2021. PMID: 34458633 Free PMC article.
-
Huntington's Disease: Complex Pathogenesis and Therapeutic Strategies.Int J Mol Sci. 2024 Mar 29;25(7):3845. doi: 10.3390/ijms25073845. Int J Mol Sci. 2024. PMID: 38612657 Free PMC article. Review.
-
Suppressing aberrant GluN3A expression rescues synaptic and behavioral impairments in Huntington's disease models.Nat Med. 2013 Aug;19(8):1030-8. doi: 10.1038/nm.3246. Epub 2013 Jul 14. Nat Med. 2013. PMID: 23852340 Free PMC article.
References
-
- Alzheimer C, Schwindt PC, Crill WE. Postnatal development of a persistent Na+ current in pyramidal neurons from rat sensorimotor cortex. J Neurophysiol. 1993;69:290–292. - PubMed
-
- André VM, Cepeda C, Venegas A, Gomez Y, Levine MS. Altered cortical glutamate receptor function in the R6/2 model of Huntington's disease. J Neurophysiol. 2006;95:2108–2119. - PubMed
-
- Ariano MA, Cepeda C, Calvert CR, Flores-Hernández J, Hernández-Echeagaray E, Klapstein GJ, Chandler SH, Aronin N, DiFiglia M, Levine MS. Striatal potassium channel dysfunction in Huntington's disease transgenic mice. J Neurophysiol. 2005;93:2565–2574. - PubMed
-
- Arning L, Kraus PH, Valentin S, Saft C, Andrich J, Epplen JT. NR2A and NR2B receptor gene variations modify age at onset in Huntington disease. Neurogenetics. 2005;6:25–28. - PubMed
-
- Arzberger T, Krampfl K, Leimgruber S, Weindl A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington's disease–an in situ hybridization study. J Neuropathol Exp Neurol. 1997;56:440–454. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases