

Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains
- PMID: 19229312
- PMCID: PMC2636890
- DOI: 10.1371/journal.ppat.1000310
Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains
Abstract
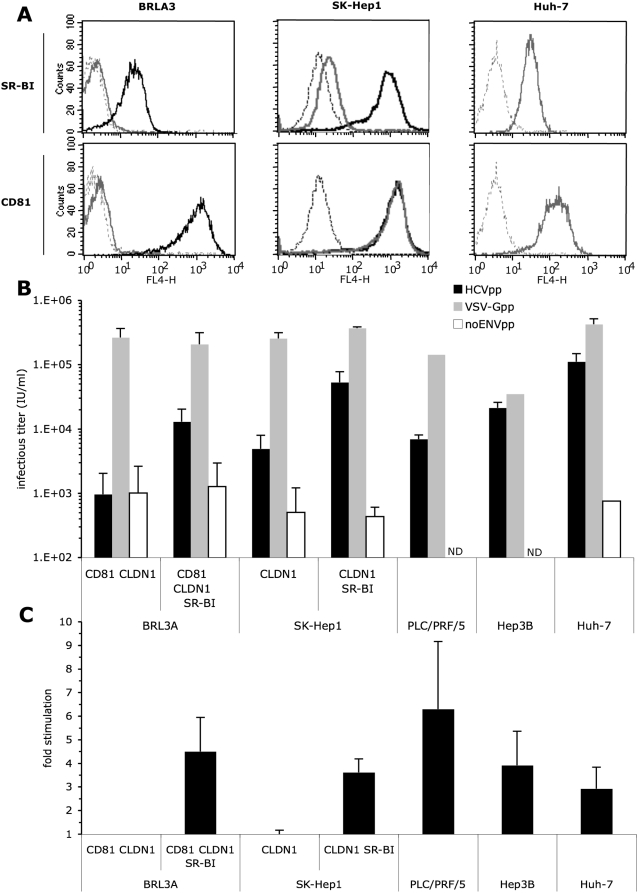
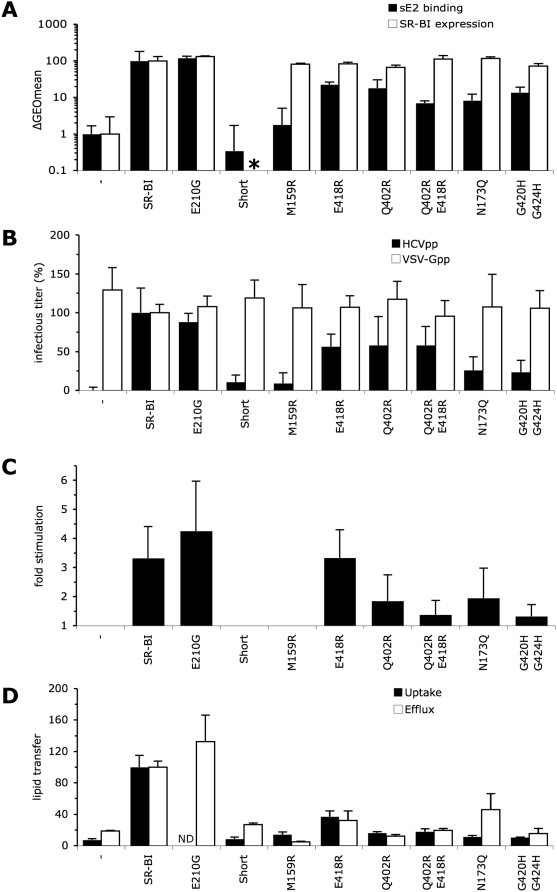
HCV entry into cells is a multi-step and slow process. It is believed that the initial capture of HCV particles by glycosaminoglycans and/or lipoprotein receptors is followed by coordinated interactions with the scavenger receptor class B type I (SR-BI), a major receptor of high-density lipoprotein (HDL), the CD81 tetraspanin, and the tight junction protein Claudin-1, ultimately leading to uptake and cellular penetration of HCV via low-pH endosomes. Several reports have indicated that HDL promotes HCV entry through interaction with SR-BI. This pathway remains largely elusive, although it was shown that HDL neither associates with HCV particles nor modulates HCV binding to SR-BI. In contrast to CD81 and Claudin-1, the importance of SR-BI has only been addressed indirectly because of lack of cells in which functional complementation assays with mutant receptors could be performed. Here we identified for the first time two cell types that supported HCVpp and HCVcc entry upon ectopic SR-BI expression. Remarkably, the undetectable expression of SR-BI in rat hepatoma cells allowed unambiguous investigation of human SR-BI functions during HCV entry. By expressing different SR-BI mutants in either cell line, our results revealed features of SR-BI intracellular domains that influence HCV infectivity without affecting receptor binding and stimulation of HCV entry induced by HDL/SR-BI interaction. Conversely, we identified positions of SR-BI ectodomain that, by altering HCV binding, inhibit entry. Finally, we characterized alternative ectodomain determinants that, by reducing SR-BI cholesterol uptake and efflux functions, abolish HDL-mediated infection-enhancement. Altogether, we demonstrate that SR-BI is an essential HCV entry factor. Moreover, our results highlight specific SR-BI determinants required during HCV entry and physiological lipid transfer functions hijacked by HCV to favor infection.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures




References
-
- Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–567. - PubMed
-
- Bartosch B, Cosset F-L. Cell entry of hepatitis C virus. Virology. 2006;348:1–12. - PubMed
-
- Dubuisson J, Helle F, Cocquerel L. Early steps of the hepatitis C virus life cycle. Cell Microbiol. 2008;10:821–827. - PubMed
-
- von Hahn T, Rice CM. Hepatitis C virus entry. J Biol Chem. 2008;283:3689–3693. - PubMed
-
- Thomssen R, Bonk S, Propfe C, Heermann KH, Kochel HG, et al. Association of hepatitis C virus in human sera with beta-lipoprotein. Med Microbiol Immunol. 1992;181:293–300. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
