

Biological convergence of cancer signatures
- PMID: 19229342
- PMCID: PMC2642727
- DOI: 10.1371/journal.pone.0004544
Biological convergence of cancer signatures
Abstract
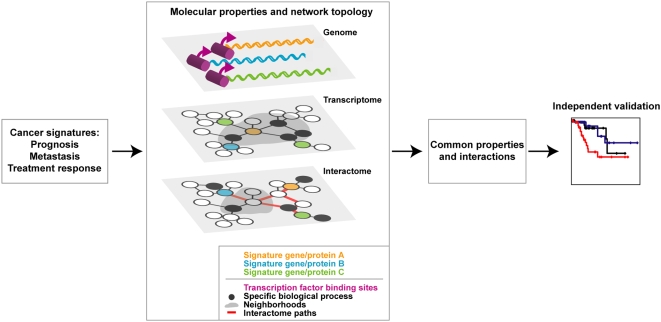
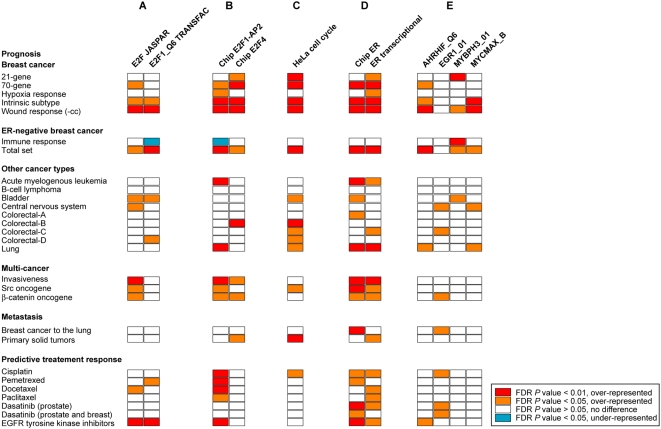
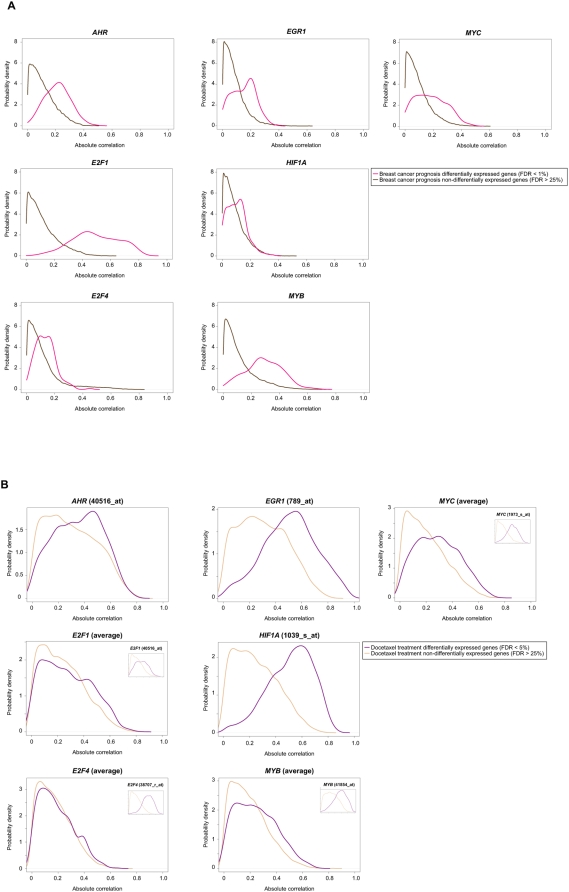
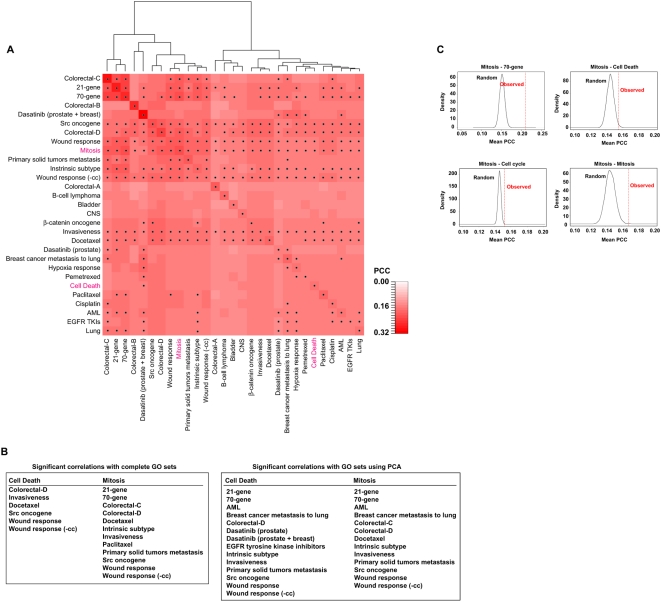
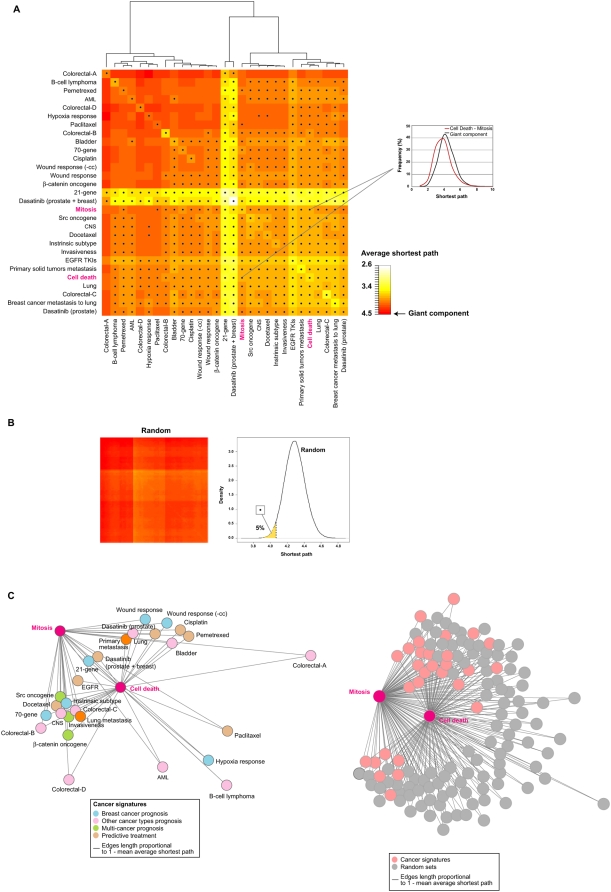
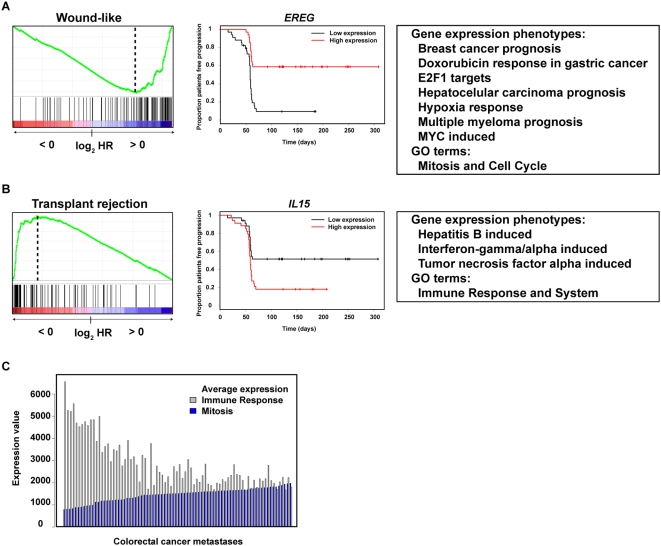
Gene expression profiling has identified cancer prognostic and predictive signatures with superior performance to conventional histopathological or clinical parameters. Consequently, signatures are being incorporated into clinical practice and will soon influence everyday decisions in oncology. However, the slight overlap in the gene identity between signatures for the same cancer type or condition raises questions about their biological and clinical implications. To clarify these issues, better understanding of the molecular properties and possible interactions underlying apparently dissimilar signatures is needed. Here, we evaluated whether the signatures of 24 independent studies are related at the genome, transcriptome or proteome levels. Significant associations were consistently observed across these molecular layers, which suggest the existence of a common cancer cell phenotype. Convergence on cell proliferation and death supports the pivotal involvement of these processes in prognosis, metastasis and treatment response. In addition, functional and molecular associations were identified with the immune response in different cancer types and conditions that complement the contribution of cell proliferation and death. Examination of additional, independent, cancer datasets corroborated our observations. This study proposes a comprehensive strategy for interpreting cancer signatures that reveals common design principles and systems-level properties.
Conflict of interest statement
Figures
Similar articles
-
Conservation of immune gene signatures in solid tumors and prognostic implications.BMC Cancer. 2016 Nov 22;16(1):911. doi: 10.1186/s12885-016-2948-z. BMC Cancer. 2016. PMID: 27871313 Free PMC article.
-
A comprehensive analysis of prognostic signatures reveals the high predictive capacity of the proliferation, immune response and RNA splicing modules in breast cancer.Breast Cancer Res. 2008;10(6):R93. doi: 10.1186/bcr2192. Epub 2008 Nov 13. Breast Cancer Res. 2008. PMID: 19014521 Free PMC article.
-
Schlafen-11 expression is associated with immune signatures and basal-like phenotype in breast cancer.Breast Cancer Res Treat. 2019 Sep;177(2):335-343. doi: 10.1007/s10549-019-05313-w. Epub 2019 Jun 20. Breast Cancer Res Treat. 2019. PMID: 31222709
-
A decade of cancer gene profiling: from molecular portraits to molecular function.Methods Mol Biol. 2010;576:61-87. doi: 10.1007/978-1-59745-545-9_5. Methods Mol Biol. 2010. PMID: 19882258 Review.
-
Gene expression profiling in breast cancer.Curr Opin Oncol. 2007 Nov;19(6):547-51. doi: 10.1097/CCO.0b013e3282f0ada3. Curr Opin Oncol. 2007. PMID: 17906450 Review.
Cited by
-
A combined oncogenic pathway signature of BRAF, KRAS and PI3KCA mutation improves colorectal cancer classification and cetuximab treatment prediction.Gut. 2013 Apr;62(4):540-9. doi: 10.1136/gutjnl-2012-302423. Epub 2012 Jul 14. Gut. 2013. PMID: 22798500 Free PMC article.
-
Cancer develops, progresses and responds to therapies through restricted perturbation of the protein-protein interaction network.Integr Biol (Camb). 2012 Sep;4(9):1038-48. doi: 10.1039/c2ib20052j. Epub 2012 Jul 18. Integr Biol (Camb). 2012. PMID: 22806580 Free PMC article.
-
Do two machine-learning based prognostic signatures for breast cancer capture the same biological processes?PLoS One. 2011 Mar 14;6(3):e17795. doi: 10.1371/journal.pone.0017795. PLoS One. 2011. PMID: 21423753 Free PMC article.
-
Clinical value of prognosis gene expression signatures in colorectal cancer: a systematic review.PLoS One. 2012;7(11):e48877. doi: 10.1371/journal.pone.0048877. Epub 2012 Nov 7. PLoS One. 2012. PMID: 23145004 Free PMC article.
-
Improving biomarker list stability by integration of biological knowledge in the learning process.BMC Bioinformatics. 2012 Mar 28;13 Suppl 4(Suppl 4):S22. doi: 10.1186/1471-2105-13-S4-S22. BMC Bioinformatics. 2012. PMID: 22536969 Free PMC article.
References
-
- Nuyten DS, van de Vijver MJ. Using microarray analysis as a prognostic and predictive tool in oncology: focus on breast cancer and normal tissue toxicity. Semin Radiat Oncol. 2008;18:105–114. - PubMed
-
- Morris SR, Carey LA. Gene expression profiling in breast cancer. Curr Opin Oncol. 2007;19:547–551. - PubMed
-
- Eden P, Ritz C, Rose C, Ferno M, Peterson C. “Good Old” clinical markers have similar power in breast cancer prognosis as microarray gene expression profilers. Eur J Cancer. 2004;40:1837–1841. - PubMed
-
- Lin YH, Friederichs J, Black MA, Mages J, Rosenberg R, et al. Multiple gene expression classifiers from different array platforms predict poor prognosis of colorectal cancer. Clin Cancer Res. 2007;13:498–507. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
