

Calcium signaling and neurodegenerative diseases
- PMID: 19230774
- PMCID: PMC3226745
- DOI: 10.1016/j.molmed.2009.01.001
Calcium signaling and neurodegenerative diseases
Abstract
Neurodegenerative disorders, such as Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD) and spinocerebellar ataxias (SCAs), present an enormous medical, social, financial and scientific problem. Recent evidence indicates that neuronal calcium (Ca2+) signaling is abnormal in many of these disorders. Similar, but less severe, changes in neuronal Ca2+ signaling occur as a result of the normal aging process. The role of aberrant neuronal Ca2+ signaling in the pathogenesis of neurodegenerative disorders is discussed here. The potential utility of Ca2+ blockers for treatment of these disorders is also highlighted. It is reasoned that Ca2+ blockers will be most beneficial clinically when used in combination with other disease-specific therapeutic approaches.
Conflict of interest statement
The author has no conflicts of interest to declare.
Figures
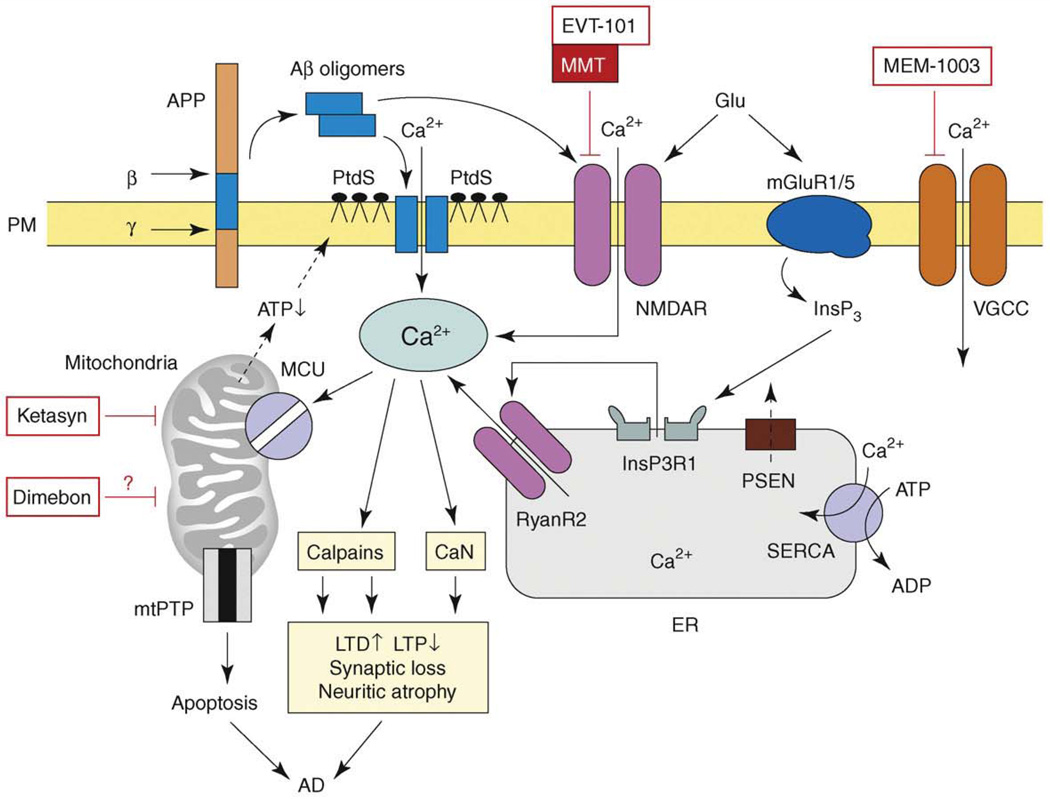
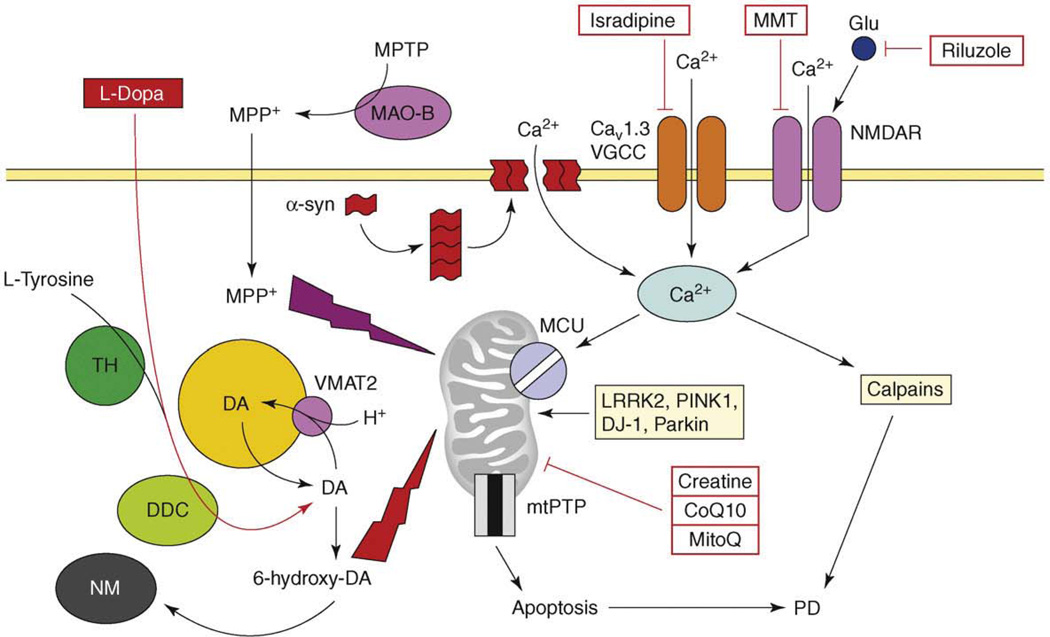
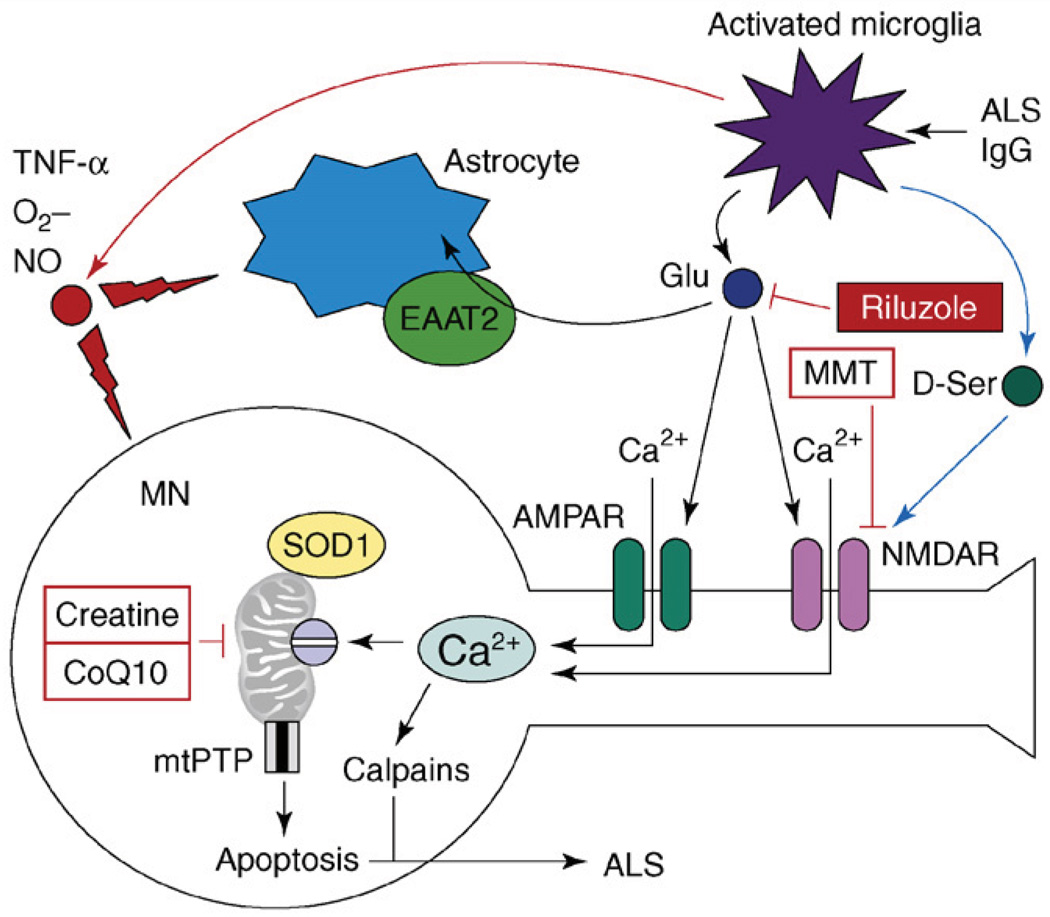
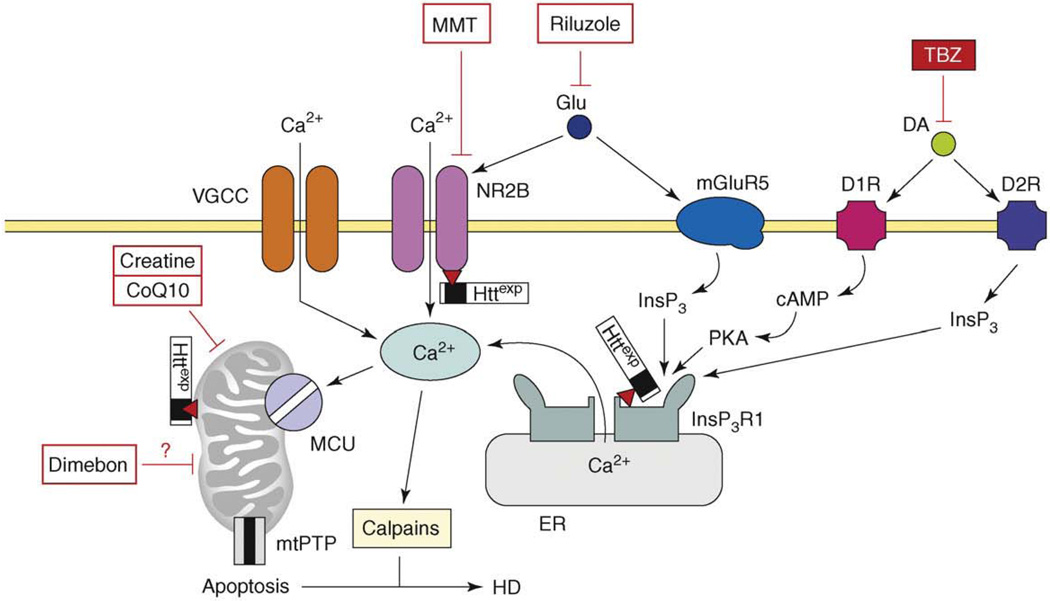
References
-
- Toescu EC, Verkhratsky A. The importance of being subtle: small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell. 2007;6:267–273. - PubMed
-
- Foster TC. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell. 2007;6:319–325. - PubMed
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. - PubMed
-
- Seabrook GR, et al. Beyond amyloid: the next generation of Alzheimer’s disease therapeutics. Mol. Interv. 2007;7:261–270. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
