

Distal Xq duplication and functional Xq disomy
- PMID: 19232094
- PMCID: PMC2649904
- DOI: 10.1186/1750-1172-4-4
Distal Xq duplication and functional Xq disomy
Abstract
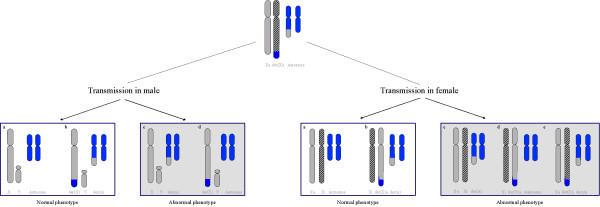
Distal Xq duplications refer to chromosomal disorders resulting from involvement of the long arm of the X chromosome (Xq). Clinical manifestations widely vary depending on the gender of the patient and on the gene content of the duplicated segment. Prevalence of Xq duplications remains unknown. About 40 cases of Xq28 functional disomy due to cytogenetically visible rearrangements, and about 50 cases of cryptic duplications encompassing the MECP2 gene have been reported. The most frequently reported distal duplications involve the Xq28 segment and yield a recognisable phenotype including distinctive facial features (premature closure of the fontanels or ridged metopic suture, broad face with full cheeks, epicanthal folds, large ears, small and open mouth, ear anomalies, pointed nose, abnormal palate and facial hypotonia), major axial hypotonia, severe developmental delay, severe feeding difficulties, abnormal genitalia and proneness to infections. Xq duplications may be caused either by an intrachromosomal duplication or an unbalanced X/Y or X/autosome translocation. In XY males, structural X disomy always results in functional disomy. In females, failure of X chromosome dosage compensation could result from a variety of mechanisms, including an unfavourable pattern of inactivation, a breakpoint separating an X segment from the X-inactivation centre in cis, or a small ring chromosome. The MECP2 gene in Xq28 is the most important dosage-sensitive gene responsible for the abnormal phenotype in duplications of distal Xq. Diagnosis is based on clinical features and is confirmed by CGH array techniques. Differential diagnoses include Prader-Willi syndrome and Alpha thalassaemia-mental retardation, X linked (ATR-X). The recurrence risk is significant if a structural rearrangement is present in one of the parent, the most frequent situation being that of an intrachromosomal duplication inherited from the mother. Prenatal diagnosis is performed by cytogenetic testing including FISH and/or DNA quantification methods. Management is multi-specialist and only symptomatic, with special attention to prevention of malnutrition and recurrent infections. Educational and rehabilitation support should be offered to all patients.
Figures

References
-
- Cheng SF, Rauen KA, Pinkel D, Albertson DG, Cotter PD. Xq chromosome duplication in males: clinical, cytogenetic and array CGH characterization of a new case and review. Am J Med Genet A. 2005;135:308–13. - PubMed
-
- Sanlaville D, Prieur M, de Blois MC, Genevieve D, Lapierre JM, Ozilou C, Picq M, Gosset P, Morichon-Delvallez N, Munnich A, Cormier-Daire V, Baujat G, Romana S, Vekemans M, Turleau C. Functional disomy of the Xq28 chromosome region. Eur J Hum Genet. 2005;13:579–85. doi: 10.1038/sj.ejhg.5201384. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
