

Corneal dystrophies
- PMID: 19236704
- PMCID: PMC2695576
- DOI: 10.1186/1750-1172-4-7
Corneal dystrophies
Abstract
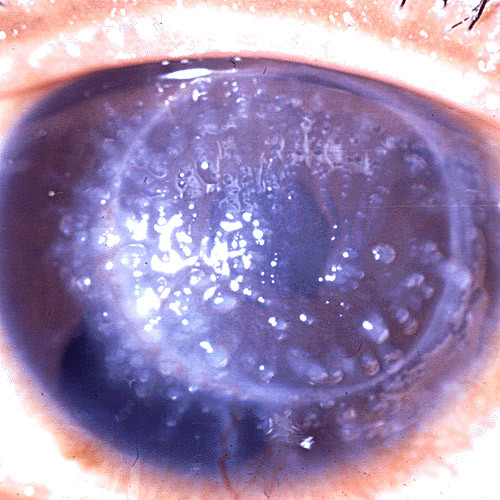
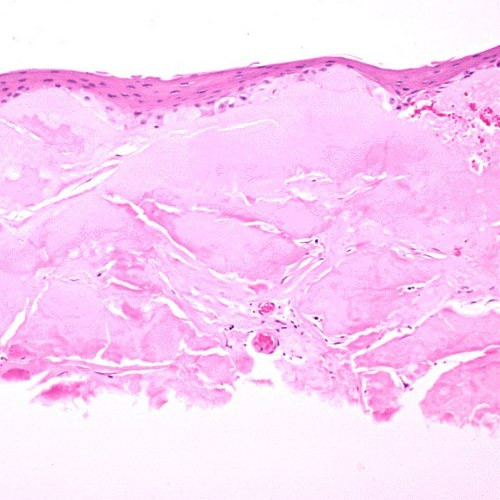
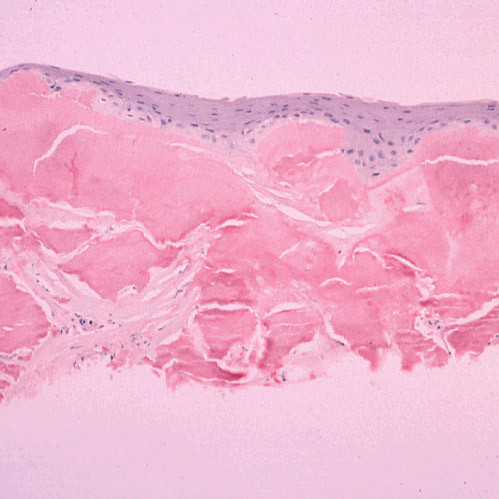
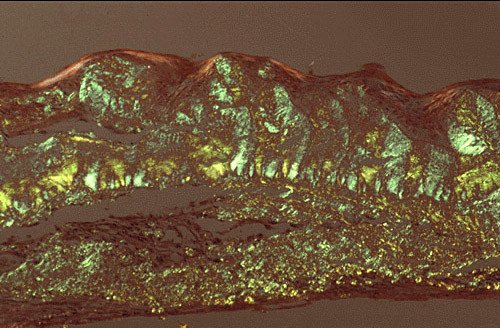
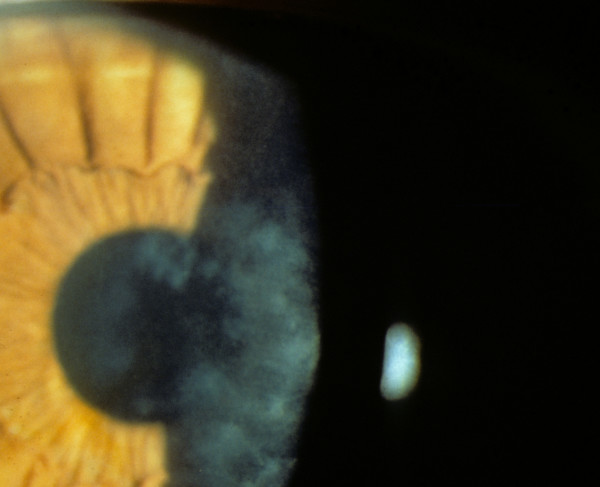
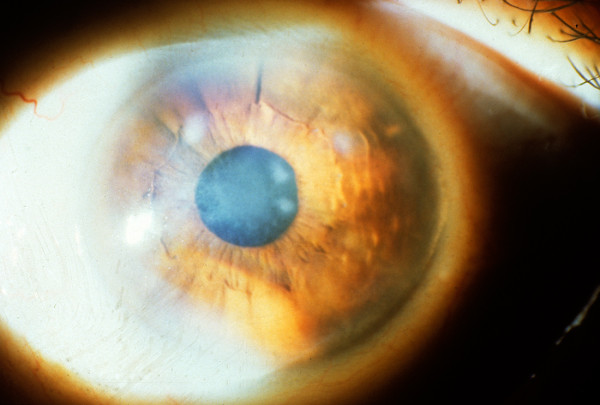
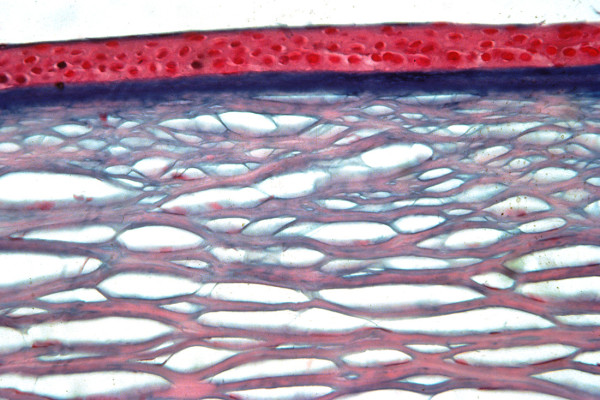
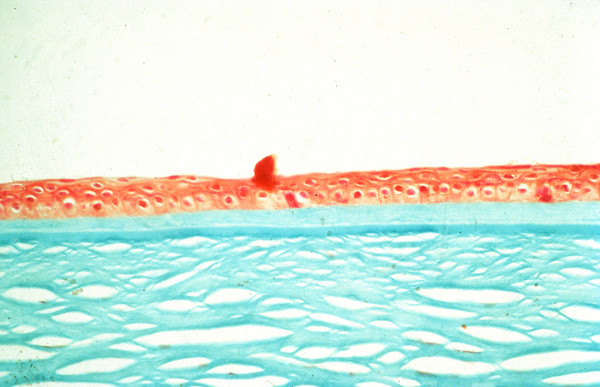
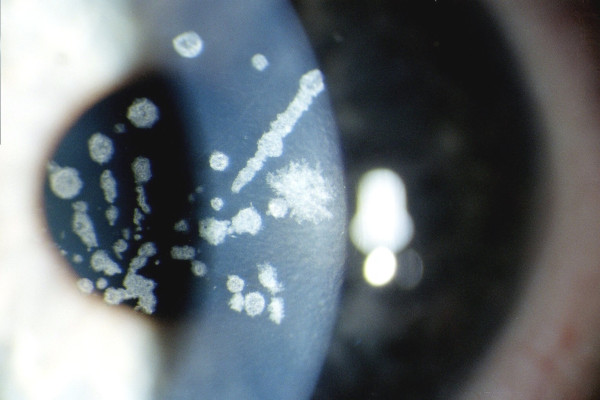
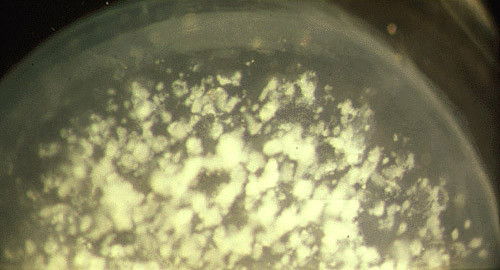
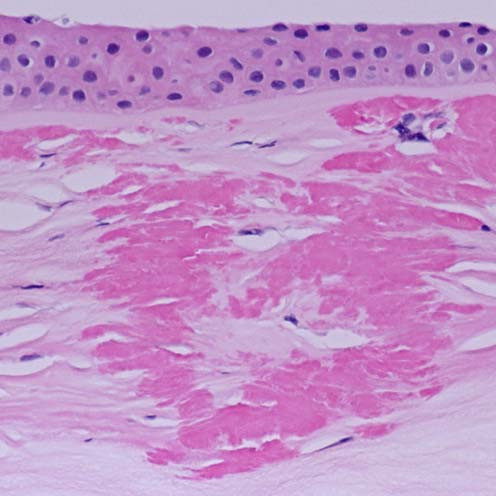
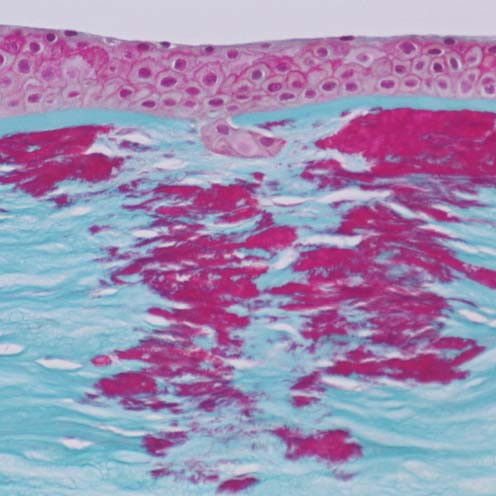
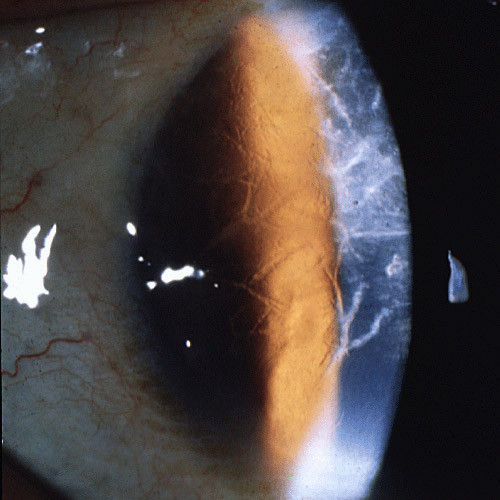
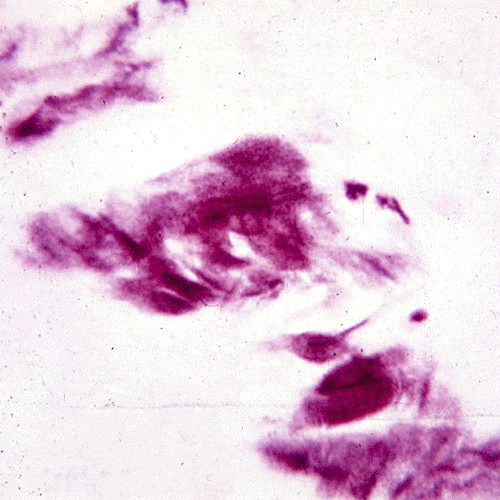
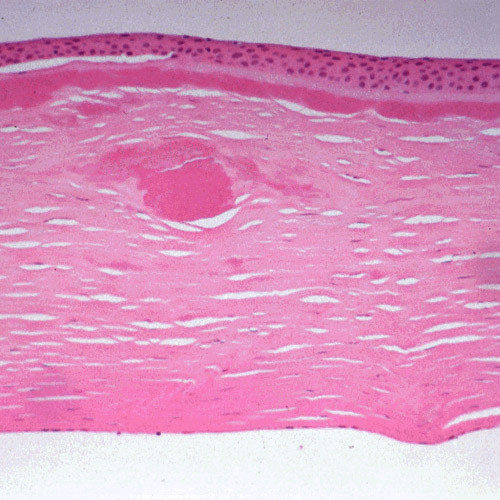
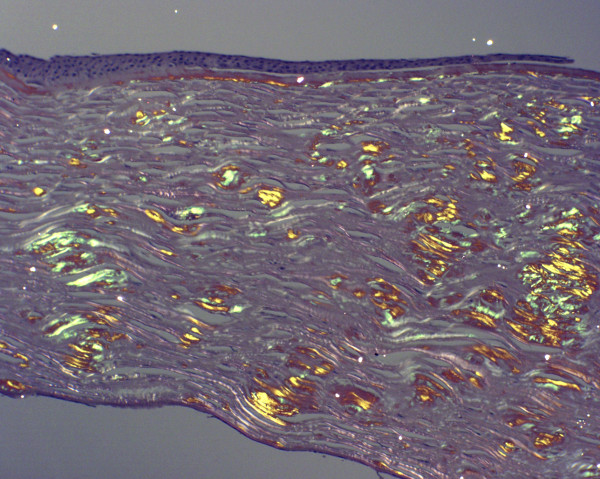
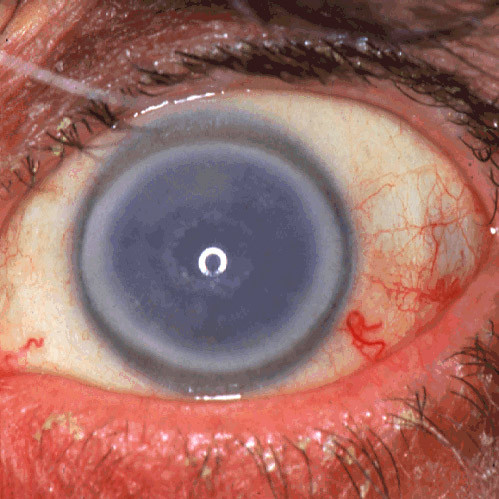
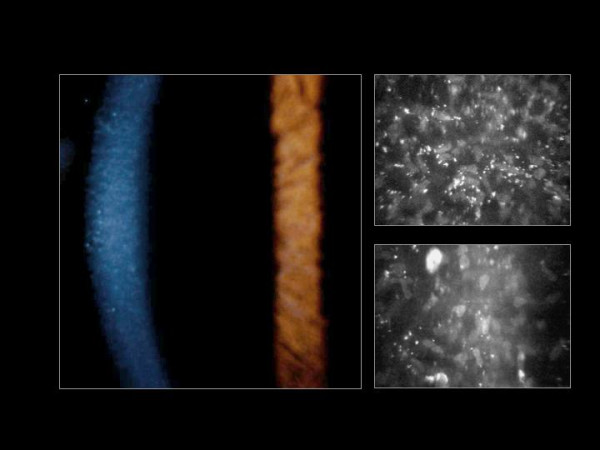
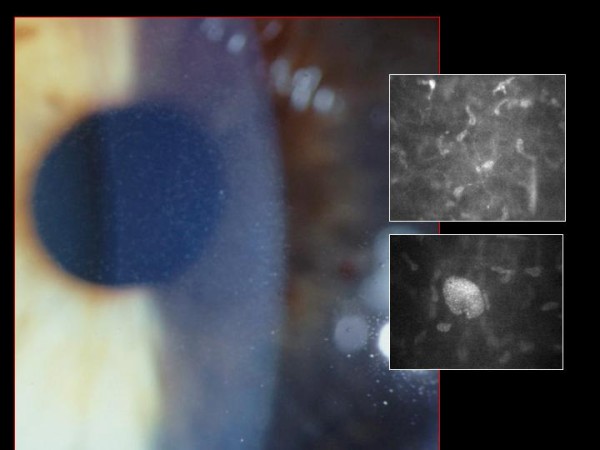
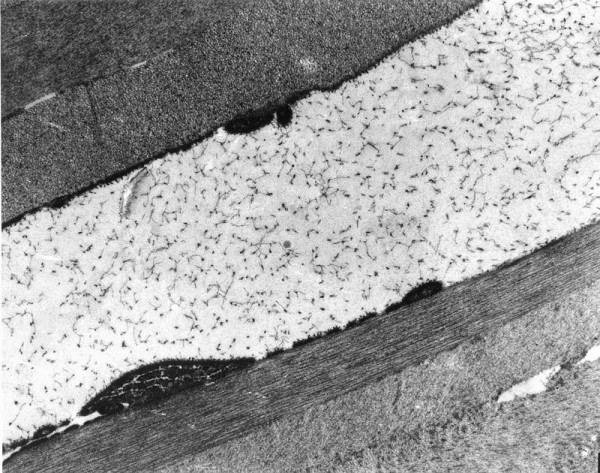
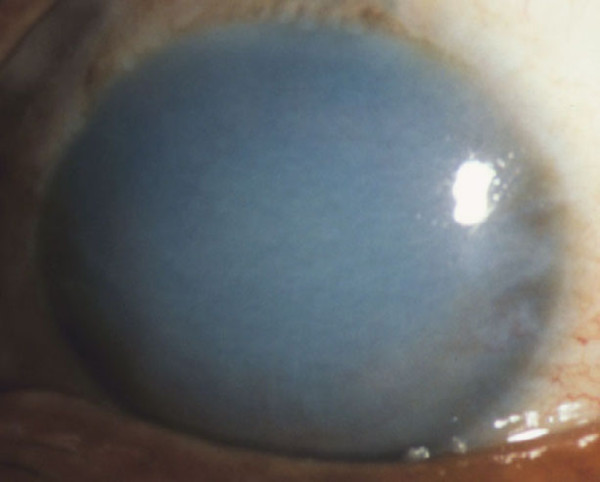
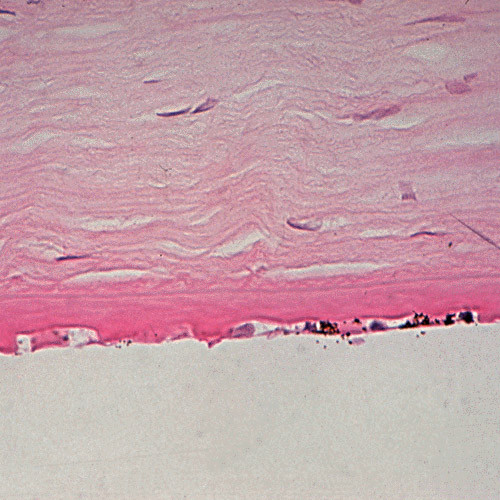
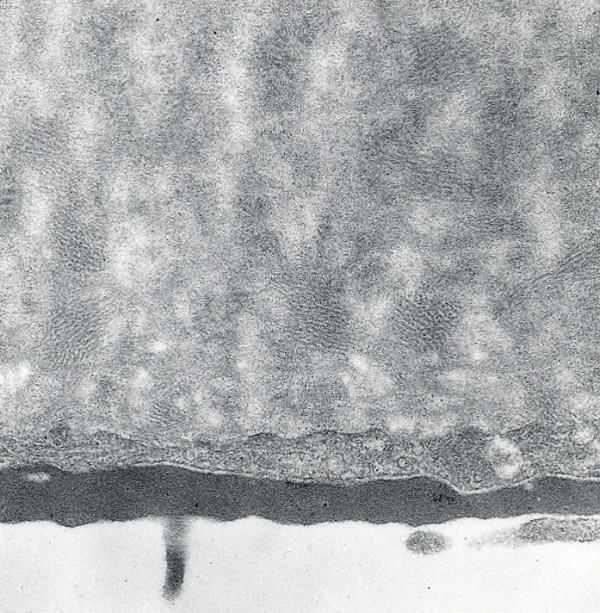
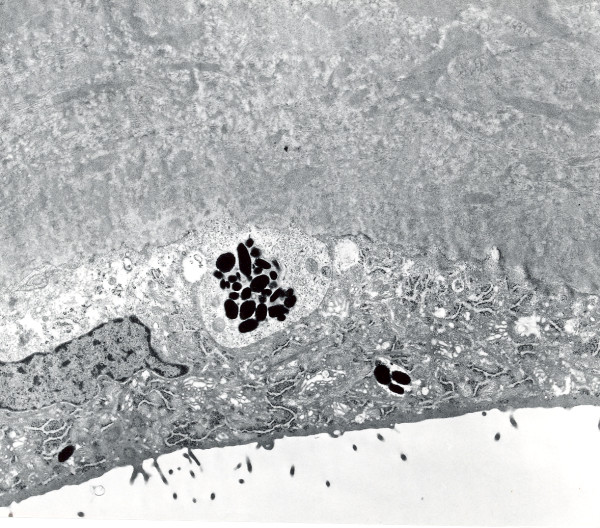
The term corneal dystrophy embraces a heterogenous group of bilateral genetically determined non-inflammatory corneal diseases that are restricted to the cornea. The designation is imprecise but remains in vogue because of its clinical value. Clinically, the corneal dystrophies can be divided into three groups based on the sole or predominant anatomical location of the abnormalities. Some affect primarily the corneal epithelium and its basement membrane or Bowman layer and the superficial corneal stroma (anterior corneal dystrophies), the corneal stroma (stromal corneal dystrophies), or Descemet membrane and the corneal endothelium (posterior corneal dystrophies). Most corneal dystrophies have no systemic manifestations and present with variable shaped corneal opacities in a clear or cloudy cornea and they affect visual acuity to different degrees. Corneal dystrophies may have a simple autosomal dominant, autosomal recessive or X-linked recessive Mendelian mode of inheritance. Different corneal dystrophies are caused by mutations in the CHST6, KRT3, KRT12, PIP5K3, SLC4A11, TACSTD2, TGFBI, and UBIAD1 genes. Knowledge about the responsible genetic mutations responsible for these disorders has led to a better understanding of their basic defect and to molecular tests for their precise diagnosis. Genes for other corneal dystrophies have been mapped to specific chromosomal loci, but have not yet been identified. As clinical manifestations widely vary with the different entities, corneal dystrophies should be suspected when corneal transparency is lost or corneal opacities occur spontaneously, particularly in both corneas, and especially in the presence of a positive family history or in the offspring of consanguineous parents. Main differential diagnoses include various causes of monoclonal gammopathy, lecithin-cholesterol-acyltransferase deficiency, Fabry disease, cystinosis, tyrosine transaminase deficiency, systemic lysosomal storage diseases (mucopolysaccharidoses, lipidoses, mucolipidoses), and several skin diseases (X-linked ichthyosis, keratosis follicularis spinolosa decalvans). The management of the corneal dystrophies varies with the specific disease. Some are treated medically or with methods that excise or ablate the abnormal corneal tissue, such as deep lamellar endothelial keratoplasty (DLEK) and phototherapeutic keratectomy (PTK). Other less debilitating or asymptomatic dystrophies do not warrant treatment. The prognosis varies from minimal effect on the vision to corneal blindness, with marked phenotypic variability.
Figures
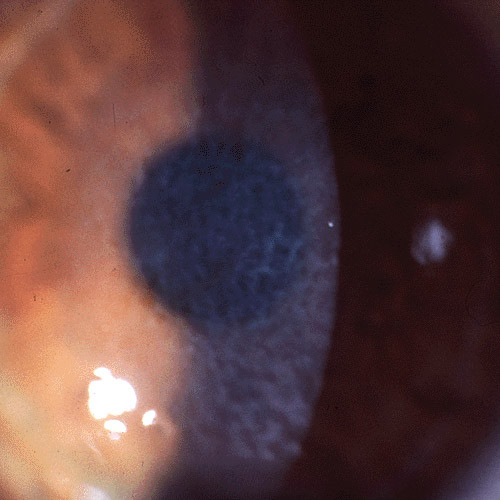
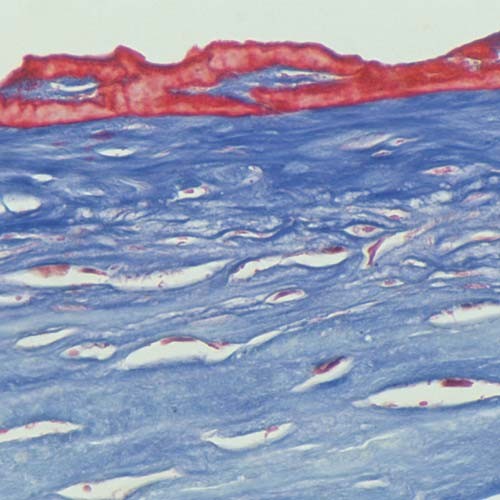
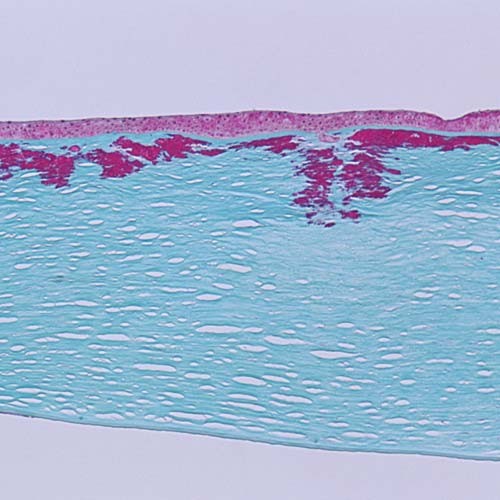
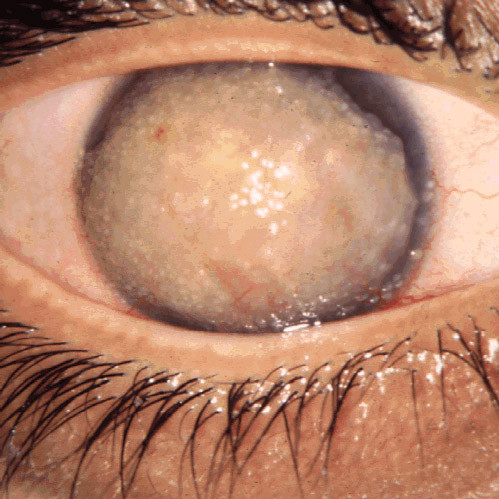

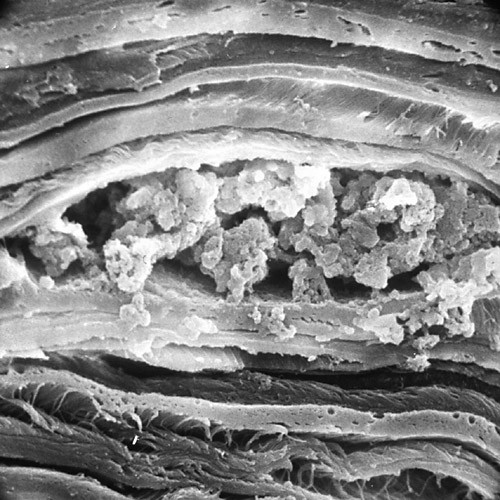
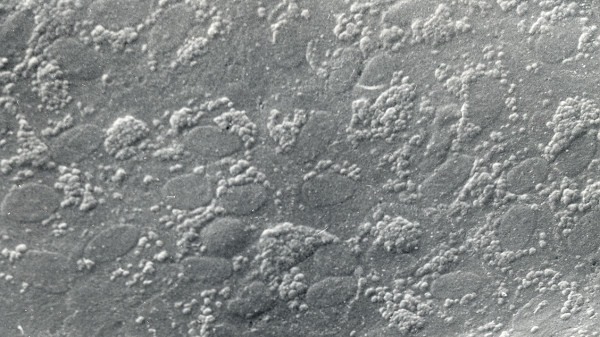
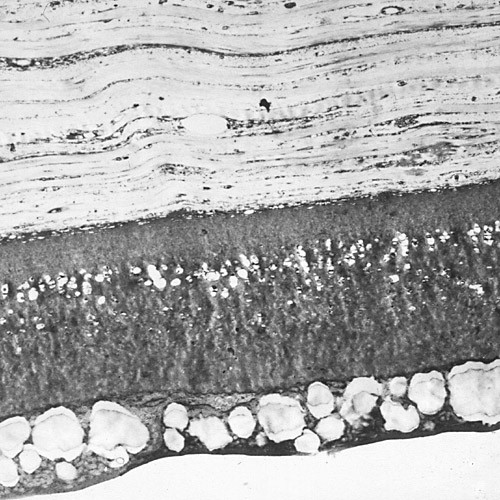

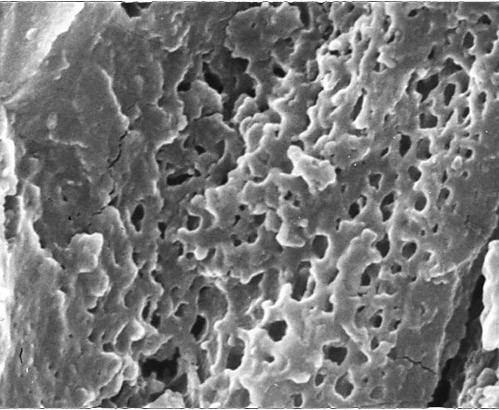
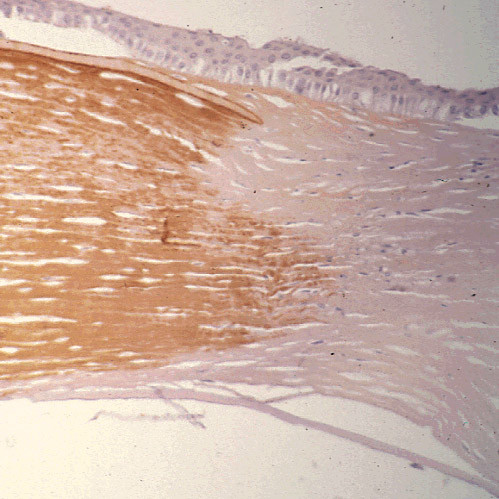
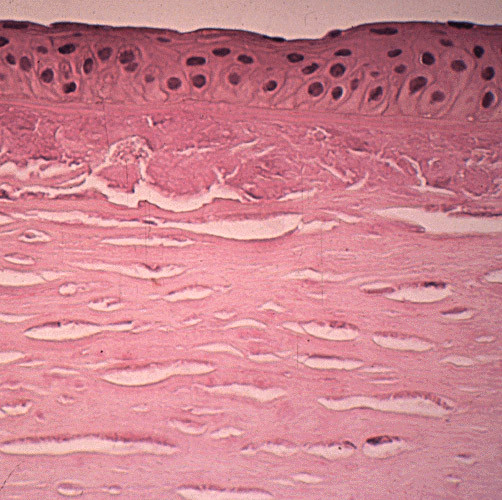
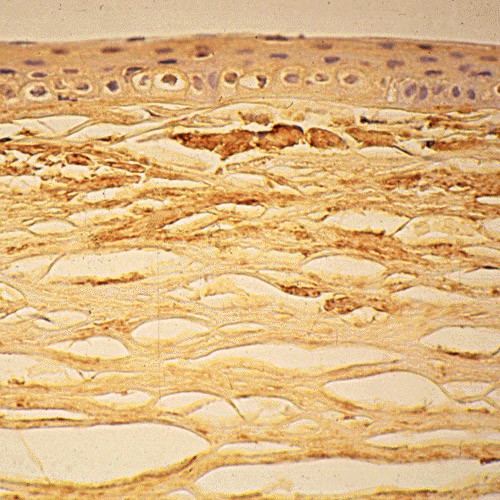
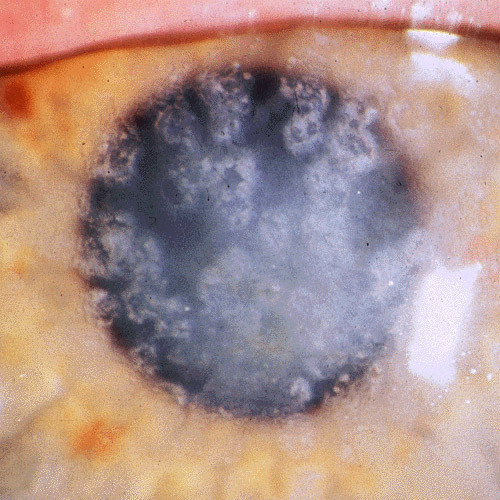
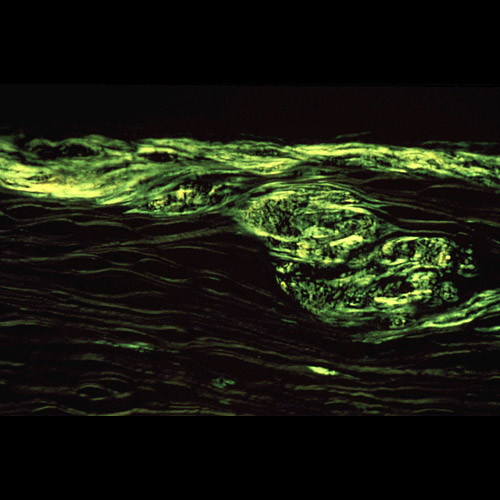
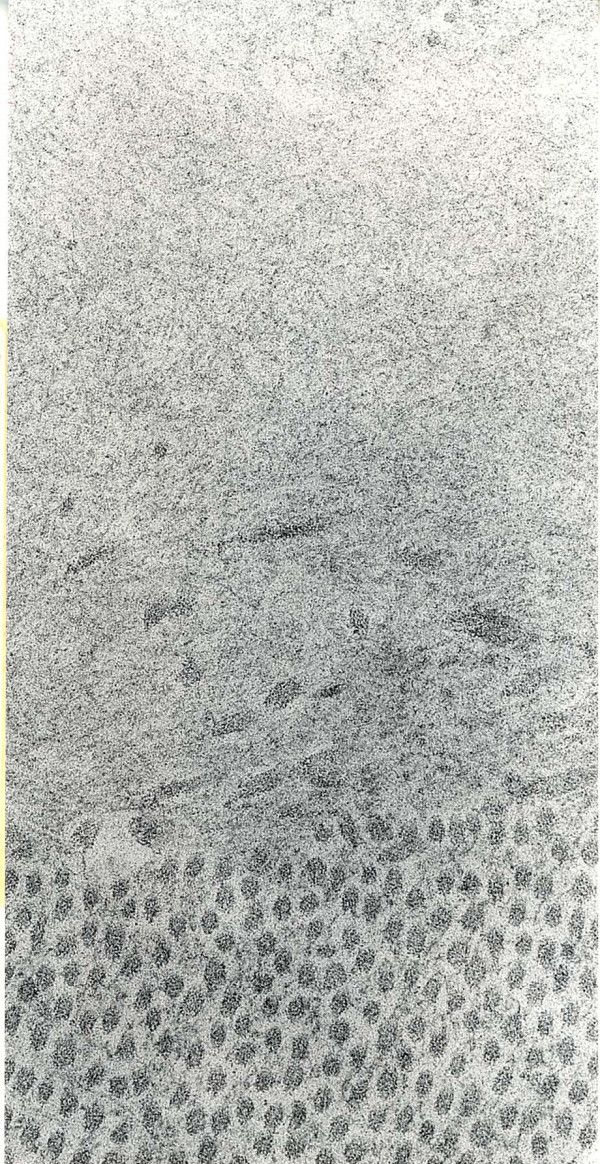

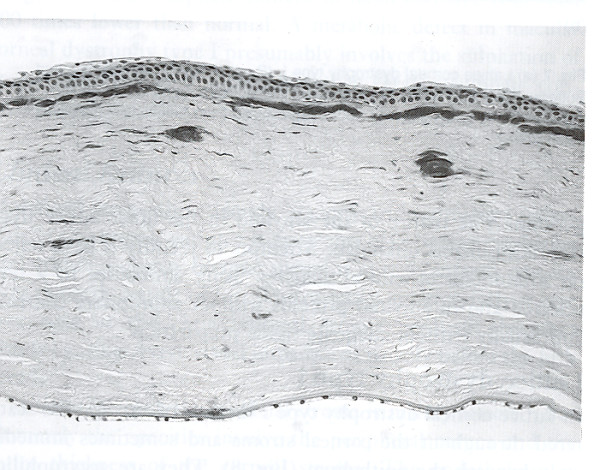
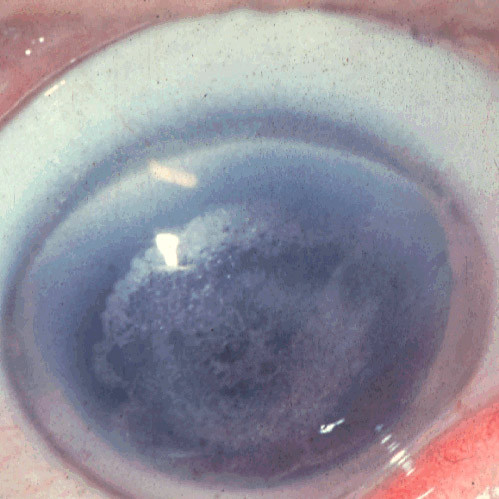



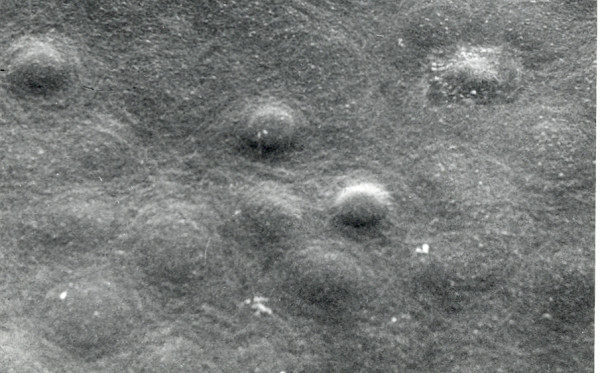

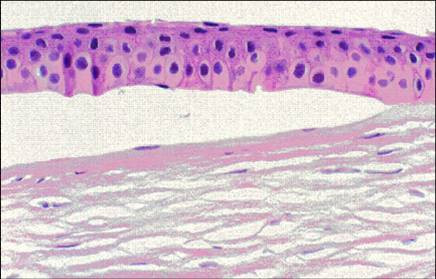
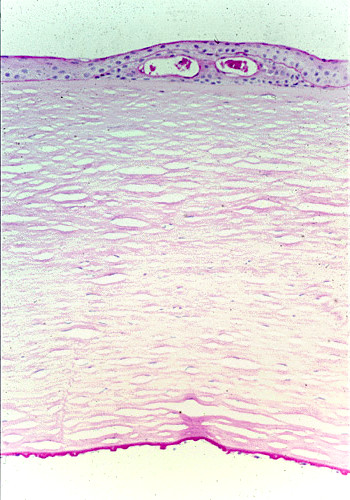
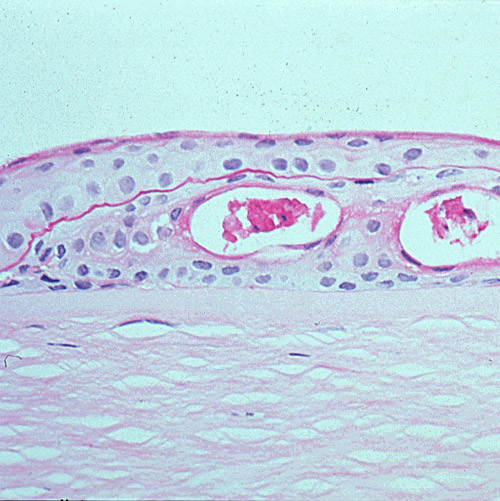
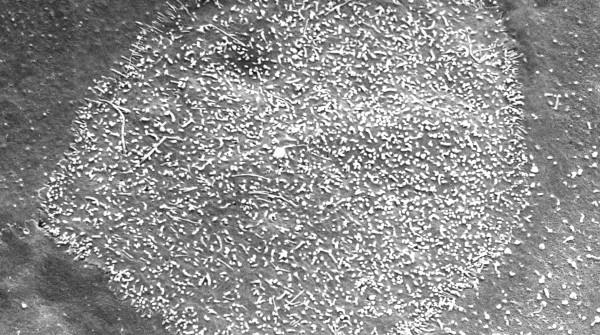
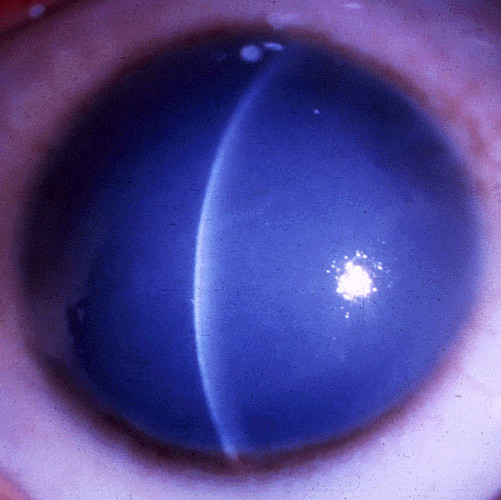


References
-
- Klintworth GK. Genetic disorders of the cornea. In: Klintworth GK, Garner A, editor. Garner and Klintworth's Pathobiology of Ocular Disease. Third. New York: Informa Heathcare; 2008. pp. 655–712.
-
- Klintworth GK. Corneal dystrophies. In: Nicholson DH, editor. Ocular Pathology Update. New York: Masson; 1980. pp. 23–54.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
