

doi: 10.1186/gb-2009-10-2-r23.
PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data
Affiliations
- PMID: 19236709
- PMCID: PMC2688268
- DOI: 10.1186/gb-2009-10-2-r23
Item in Clipboard
PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data
Genome Biol.
.
Abstract
Personal-genomics endeavors, such as the 1000 Genomes project, are generating maps of genomic structural variants by analyzing ends of massively sequenced genome fragments. To process these we developed Paired-End Mapper (PEMer; http://sv.gersteinlab.org/pemer). This comprises an analysis pipeline, compatible with several next-generation sequencing platforms; simulation-based error models, yielding confidence-values for each structural variant; and a back-end database. The simulations demonstrated high structural variant reconstruction efficiency for PEMer's coverage-adjusted multi-cutoff scoring-strategy and showed its relative insensitivity to base-calling errors.
Figures
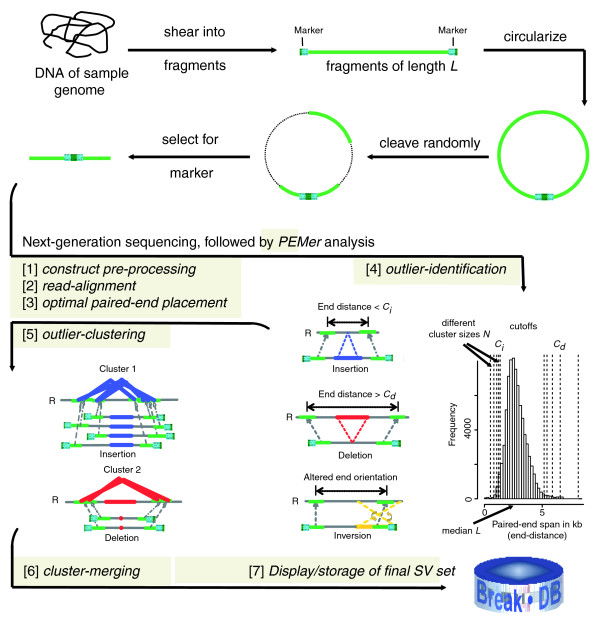
Scheme depicting computational steps carried out by PEMer. In PEM, when using the 454/Roche platform, randomly sheared genomic fragments are circularized and cleaved randomly into sequence stretches amenable to ultrafast sequencing (figure adapted and extended from Figure 1 in [21]). We subject resulting DNA sequences to PEMer for calling SVs relative to the reference genome ('R'). By default, PEMer uses the following processing steps: [1] construct pre-processing, [2] read-alignment, [3] optimal paired-end placement, [4] outlier-identification, [5] outlier-clustering, and [6] cluster-merging. Subsequently, [7] SVs (insertions, deletions, inversions, and more complex events) are displayed and stored in a back-end database for further analysis. In the outlier identification step, several different cutoff points Ci and Cd for the paired-end span, which are derived from the known insert-size distribution, are applied using a multi-cutoff strategy together with distinct minimally required paired-end cluster sizes N. After merging clusters constructed using different cutoff points, different PEM libraries, or different next-generation DNA sequencing platforms, an enhanced SV call resolution may be achieved.

Depiction of the strategy used for assigning genomic coordinates to complex SV events. Coordinates within our database BreakDB are stored in a recursive fashion, if multiple SVs with partially overlapping coordinates occurred within a single haplotype. In particular, where a coordinate is typically defined with respect to the reference genome, it can also be defined in respect to other SVs, as indicated in the scheme depicted in the figure. For example, an insertion event can take place within an earlier insertion event, affecting the same haplotype the earlier event occurred in. If coordinates for the second insertion event were reported merely relative to the human reference genome, positional information for the SV would be lost. BreakDB therefore reports both coordinates within the ancestral ('parent') event, but can also trace back all the way to the reference coordinates.
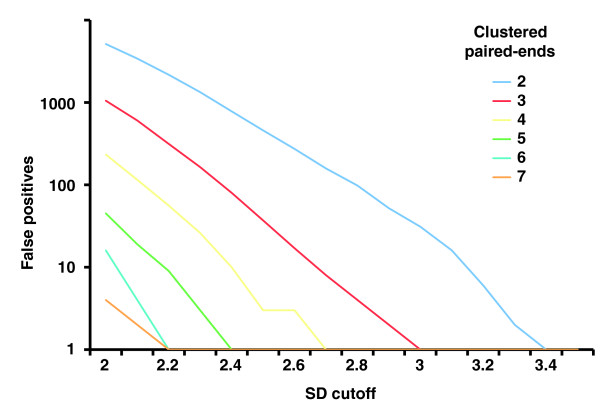
Numbers of false positive SV calls in relation to the cutoff used for defining outliers. Cutoff values for defining outlier paired ends are given in terms of standard deviations (SDs) from the median of the expected distribution of paired-end spans (which in turn is derived from the insert size). PEM data generated with the 454/Roche platform were simulated applying a median insert size L = 2.5 kb and a span-coverage of λ = 5× of the diploid chromosome 2. To arrive at λ = 5×, only optimally (uniquely) placed paired ends were considered when estimating λ ('effective span coverage'). Here, the genome-wide count of false positives is put in relation to outlier-identification cutoffs for various required cluster sizes N ('clustered paired ends') of 2 up to 7. 'False positives' refers to the number of false positives identified on chromosome 2.
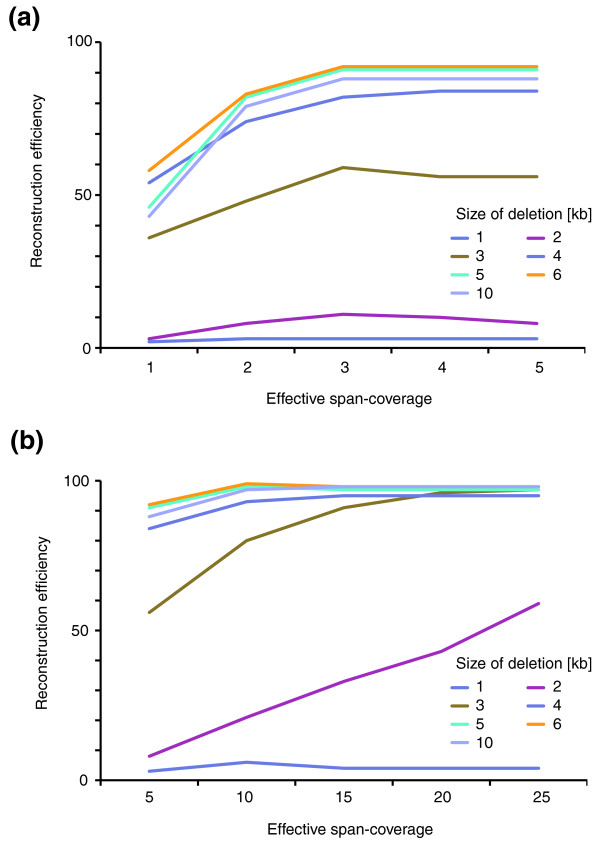
SV reconstruction efficiency in relation to the span coverage λ. We used simulations to generate heterozygous deletions 1-10 kb in size (median PEM insert size 2.5 kb; 454/Roche platform). Paired-end data were simulated with different span coverages λ; only optimally (uniquely) placed paired ends were considered when estimating λ. (a) Reconstruction efficiency for values of λ from 1× to 5×. (b) Reconstruction efficiency for values of λ from 5× to 25×.
References
-
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. - DOI - PMC - PubMed
-
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, Tyler-Smith C, Carter N, Scherer SW, Tavare S, Deloukas P, Hurles ME, Dermitzakis ET. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
