

Mimicking the folding pathway to improve homology-free protein structure prediction
- PMID: 19237560
- PMCID: PMC2656149
- DOI: 10.1073/pnas.0811363106
Mimicking the folding pathway to improve homology-free protein structure prediction
Abstract
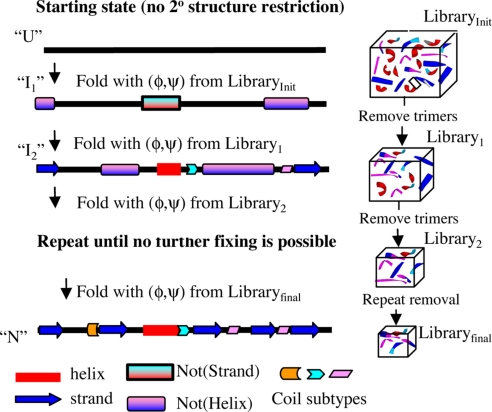
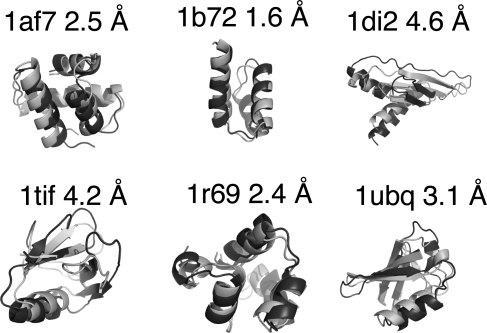
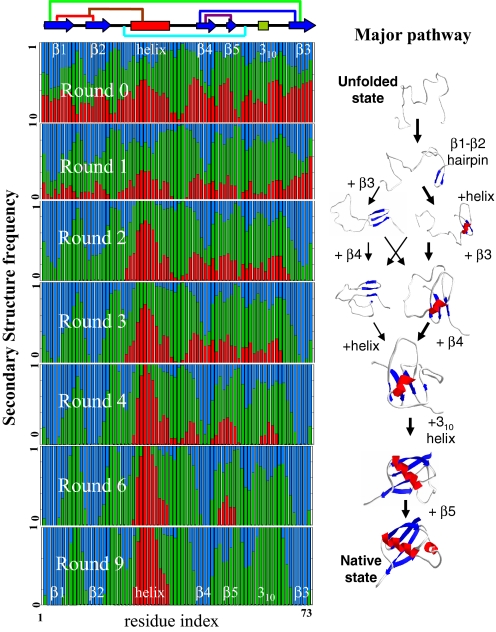
Since the demonstration that the sequence of a protein encodes its structure, the prediction of structure from sequence remains an outstanding problem that impacts numerous scientific disciplines, including many genome projects. By iteratively fixing secondary structure assignments of residues during Monte Carlo simulations of folding, our coarse-grained model without information concerning homology or explicit side chains can outperform current homology-based secondary structure prediction methods for many proteins. The computationally rapid algorithm using only single (phi,psi) dihedral angle moves also generates tertiary structures of accuracy comparable with existing all-atom methods for many small proteins, particularly those with low homology. Hence, given appropriate search strategies and scoring functions, reduced representations can be used for accurately predicting secondary structure and providing 3D structures, thereby increasing the size of proteins approachable by homology-free methods and the accuracy of template methods that depend on a high-quality input secondary structure.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Service RF. Problem solved* (*sort of) Science. 2008;321:784–786. - PubMed
-
- Pollastri G, Przybylski D, Rost B, Baldi P. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins. 2002;47:228–235. - PubMed
-
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
