

Calpain hydrolysis of alpha- and beta2-adaptins decreases clathrin-dependent endocytosis and may promote neurodegeneration
- PMID: 19240038
- PMCID: PMC2673311
- DOI: 10.1074/jbc.M804740200
Calpain hydrolysis of alpha- and beta2-adaptins decreases clathrin-dependent endocytosis and may promote neurodegeneration
Abstract
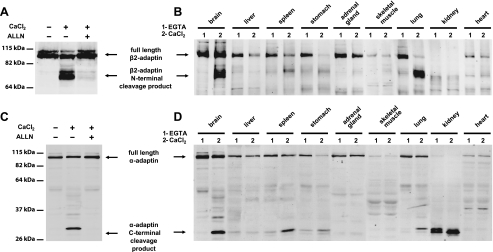
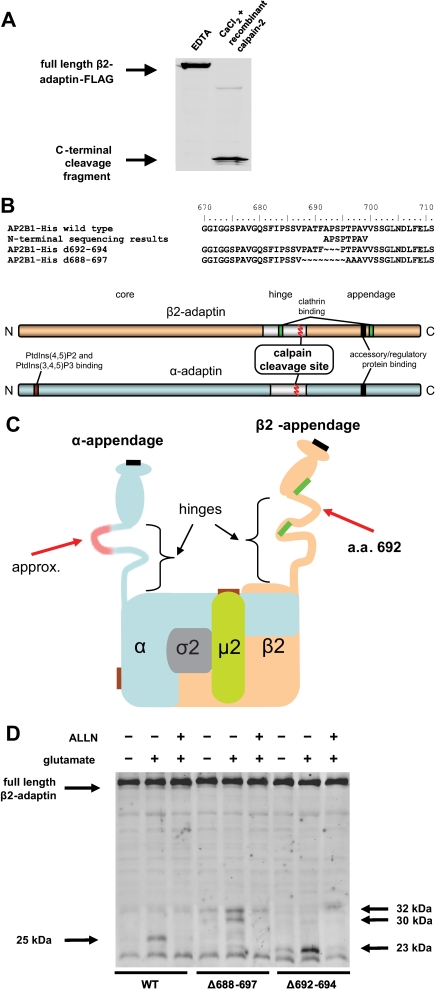
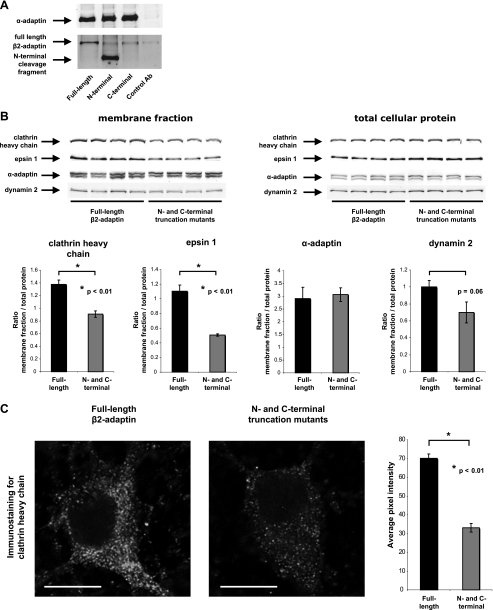
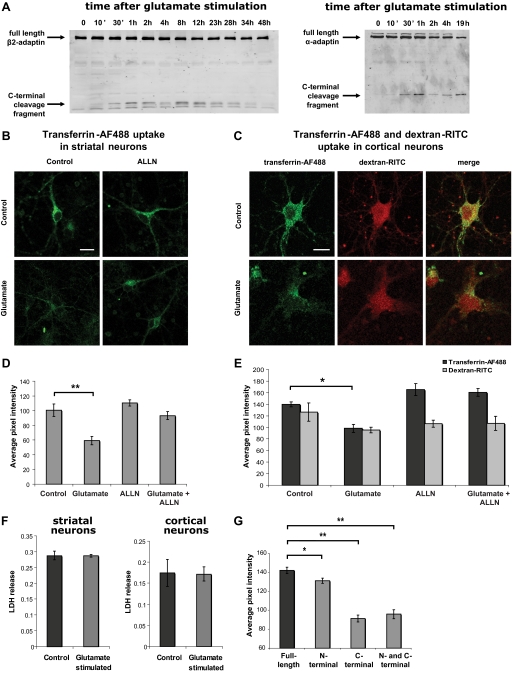
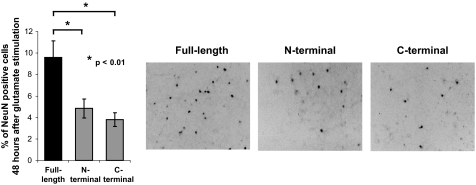
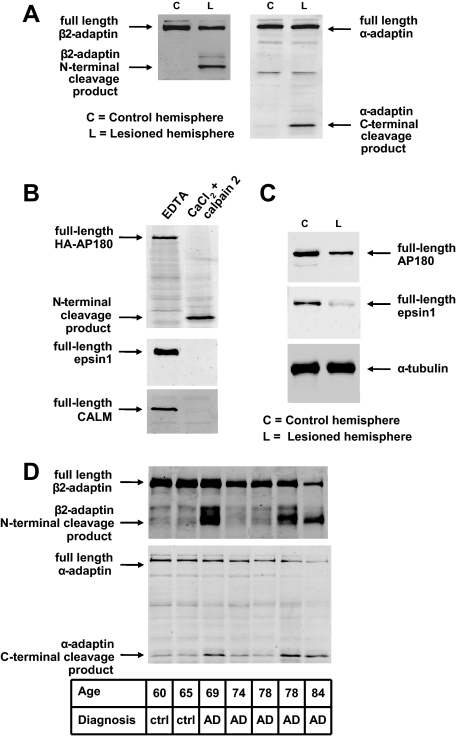
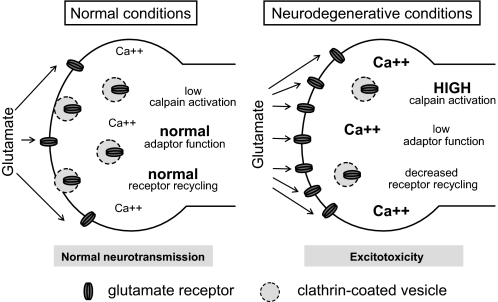
Clathrin-dependent endocytosis is mediated by a tightly regulated network of molecular interactions that provides essential protein-protein and protein-lipid binding activities. Here we report the hydrolysis of the alpha- and beta2-subunits of the tetrameric adaptor protein complex 2 by calpain. Calcium-dependent alpha- and beta2-adaptin hydrolysis was observed in several rat tissues, including brain and primary neuronal cultures. Neuronal alpha- and beta2-adaptin cleavage was inducible by glutamate stimulation and was accompanied by the decreased endocytosis of transferrin. Heterologous expression of truncated forms of the beta2-adaptin subunit significantly decreased the membrane recruitment of clathrin and inhibited clathrin-mediated receptor endocytosis. Moreover, the presence of truncated beta2-adaptin sensitized neurons to glutamate receptor-mediated excitotoxicity. Proteolysis of alpha- and beta2-adaptins, as well as the accessory clathrin adaptors epsin 1, adaptor protein 180, and the clathrin assembly lymphoid myeloid leukemia protein, was detected in brain tissues after experimentally induced ischemia and in cases of human Alzheimer disease. The present study further clarifies the central role of calpain in regulating clathrin-dependent endocytosis and provides evidence for a novel mechanism through which calpain activation may promote neurodegeneration: the sensitization of cells to glutamate-mediated excitotoxicity via the decreased internalization of surface receptors.
Figures

Similar articles
-
The beta2-adrenergic receptor/betaarrestin complex recruits the clathrin adaptor AP-2 during endocytosis.Proc Natl Acad Sci U S A. 1999 Mar 30;96(7):3712-7. doi: 10.1073/pnas.96.7.3712. Proc Natl Acad Sci U S A. 1999. PMID: 10097102 Free PMC article.
-
A single β adaptin contributes to AP1 and AP2 complexes and clathrin function in Dictyostelium.Traffic. 2012 Feb;13(2):305-16. doi: 10.1111/j.1600-0854.2011.01310.x. Epub 2011 Dec 4. Traffic. 2012. PMID: 22050483 Free PMC article.
-
Characterization of a protein phosphatase 2A holoenzyme that dephosphorylates the clathrin adaptors AP-1 and AP-2.J Biol Chem. 2008 Feb 29;283(9):5510-7. doi: 10.1074/jbc.M707166200. Epub 2007 Dec 24. J Biol Chem. 2008. PMID: 18158287
-
Clathrin and adaptors.Biochim Biophys Acta. 1998 Aug 14;1404(1-2):173-93. doi: 10.1016/s0167-4889(98)00056-1. Biochim Biophys Acta. 1998. PMID: 9714795 Review.
-
ENTH/ANTH proteins and clathrin-mediated membrane budding.J Cell Sci. 2004 Jan 1;117(Pt 1):9-18. doi: 10.1242/jcs.00928. J Cell Sci. 2004. PMID: 14657269 Review.
Cited by
-
Necrotic cell death and neurodegeneration: The involvement of endocytosis and intracellular trafficking.Worm. 2012 Jul 1;1(3):176-81. doi: 10.4161/worm.20457. Worm. 2012. PMID: 24058844 Free PMC article.
-
The Autophagy Machinery: A New Player in Chemotactic Cell Migration.Front Neurosci. 2017 Feb 16;11:78. doi: 10.3389/fnins.2017.00078. eCollection 2017. Front Neurosci. 2017. PMID: 28261054 Free PMC article. Review.
-
Inhibiting calpain 1 and 2 in cyclin G associated kinase-knockout mice mitigates podocyte injury.JCI Insight. 2020 Nov 19;5(22):e142740. doi: 10.1172/jci.insight.142740. JCI Insight. 2020. PMID: 33208557 Free PMC article.
-
Neuroprotective effect of grape seed extract on brain ischemia: a proteomic approach.Metab Brain Dis. 2019 Jun;34(3):889-907. doi: 10.1007/s11011-019-00396-2. Epub 2019 Feb 22. Metab Brain Dis. 2019. PMID: 30796716
-
Alpha adaptins show isoform-specific association with neurofibrillary tangles in Alzheimer's disease.Neuropathol Appl Neurobiol. 2022 Feb;48(2):e12776. doi: 10.1111/nan.12776. Epub 2021 Dec 7. Neuropathol Appl Neurobiol. 2022. PMID: 34820873 Free PMC article.
References
-
- Kirchhausen, T. (2000) Annu. Rev. Biochem. 69 699-727 - PubMed
-
- Schmid, E. M., and McMahon, H. T. (2007) Nature 448 883-888 - PubMed
-
- Collins, B. M., McCoy, A. J., Kent, H. M., Evans, P. R., and Owen, D. J. (2002) Cell 109 523-535 - PubMed
-
- Robinson, M. S. (2004) Trends Cell Biol. 14 167-174 - PubMed
-
- Honing, S., Ricotta, D., Krauss, M., Spate, K., Spolaore, B., Motley, A., Robinson, M., Robinson, C., Haucke, V., and Owen, D. J. (2005) Mol. Cell 18 519-531 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
