

Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington's disease mice
- PMID: 19240687
- PMCID: PMC2835182
- DOI: 10.1038/mt.2009.17
Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington's disease mice
Abstract
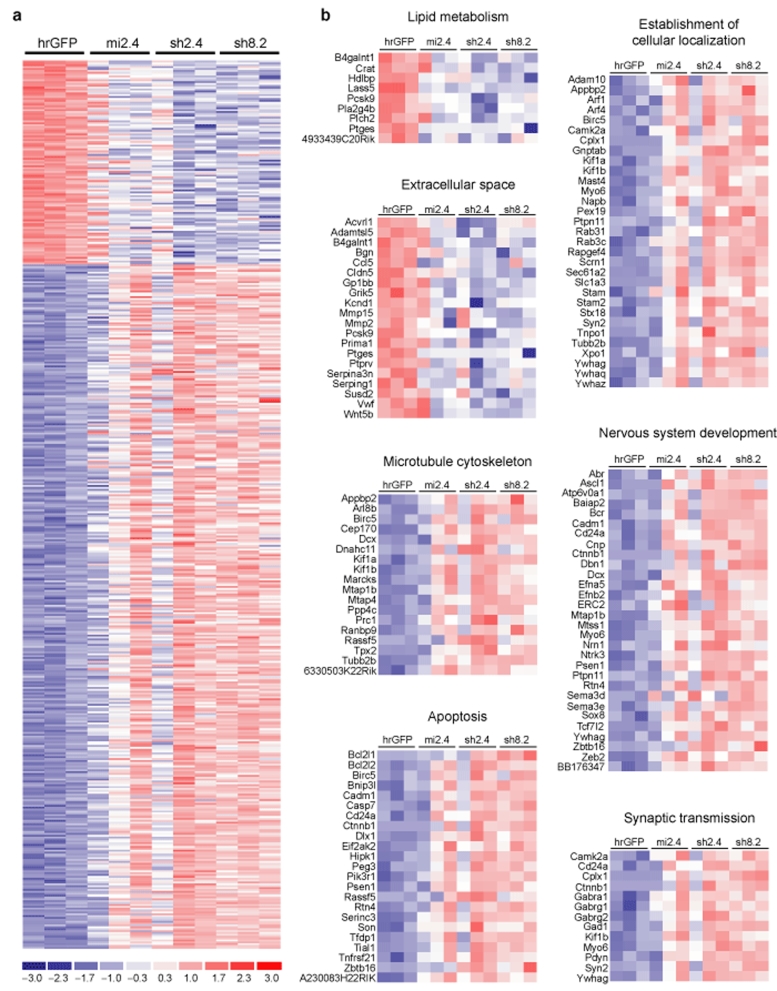
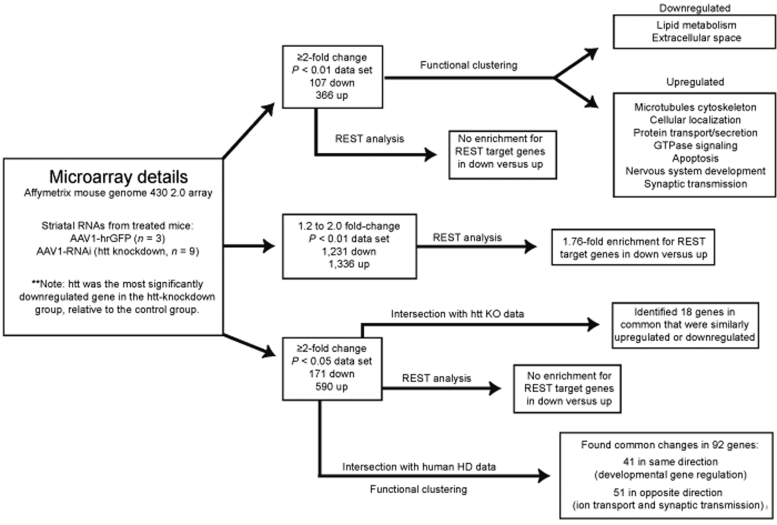
Huntington's disease (HD) is a fatal neurodegenerative disease caused by mutant huntingtin (htt) protein, and there are currently no effective treatments. Recently, we and others demonstrated that silencing mutant htt via RNA interference (RNAi) provides therapeutic benefit in HD mice. We have since found that silencing wild-type htt in adult mouse striatum is tolerated for at least 4 months. However, given the role of htt in various cellular processes, it remains unknown whether nonallele-specific silencing of both wild-type and mutant htt is a viable therapeutic strategy for HD. Here, we tested whether cosilencing wild-type and mutant htt provides therapeutic benefit and is tolerable in HD mice. After treatment, HD mice showed significant reductions in wild-type and mutant htt, and demonstrated improved motor coordination and survival. We performed transcriptional profiling to evaluate the effects of reducing wild-type htt in adult mouse striatum. We identified gene expression changes that are concordant with previously described roles for htt in various cellular processes. Also, several abnormally expressed transcripts associated with early-stage HD were differentially expressed in our studies, but intriguingly, those involved in neuronal function changed in opposing directions. Together, these encouraging and surprising findings support further testing of nonallele-specific RNAi therapeutics for HD.
Figures
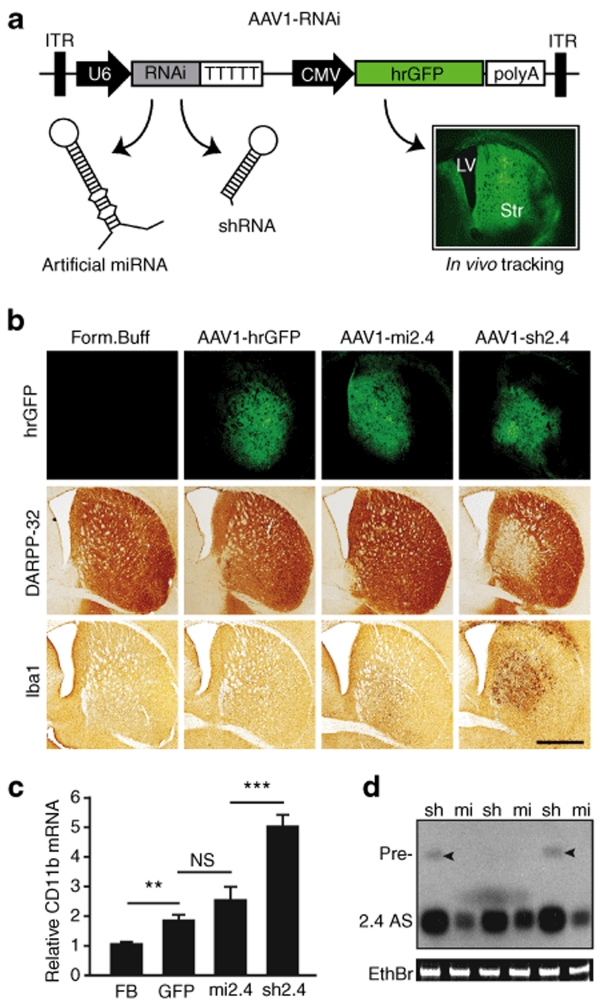
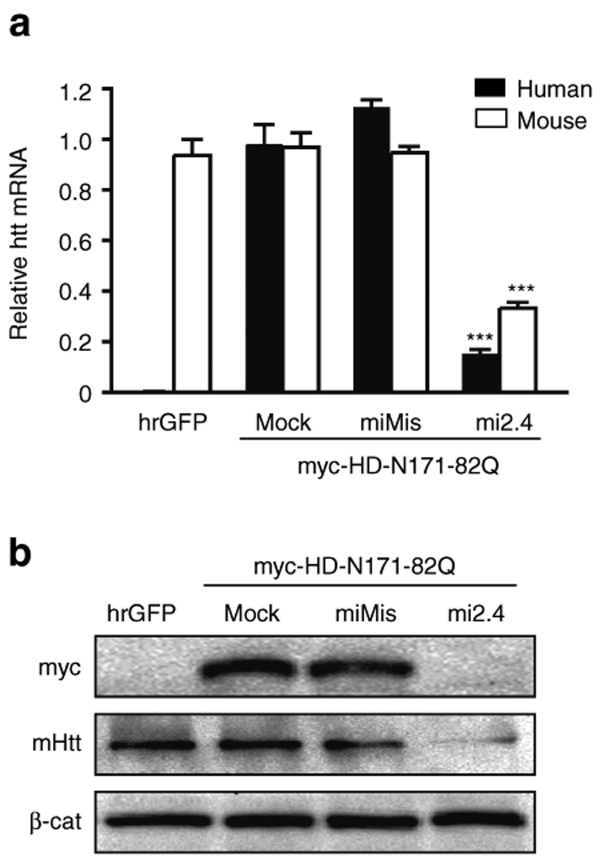
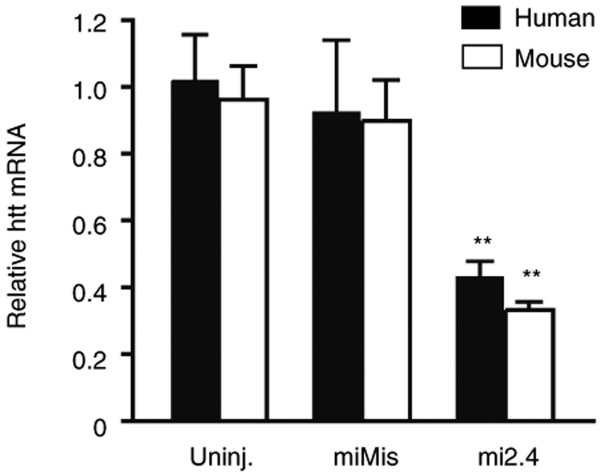
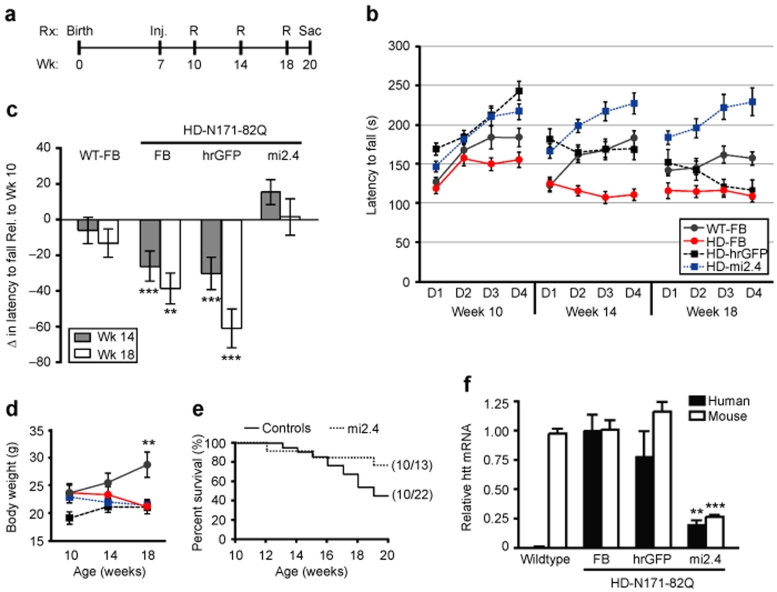
References
-
- Schaffar G, Breuer P, Boteva R, Behrends C, Tzvetkov N, Strippel N, et al. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell. 2004;15:95–105. - PubMed
-
- Saudou F, Finkbeiner S, Devys D., and , Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. - PubMed
-
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. - PubMed
-
- Zhai W, Jeong H, Cui L, Krainc D., and , Tjian R. In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell. 2005;123:1241–1253. - PubMed
-
- Qiu Z, Norflus F, Singh B, Swindell MK, Buzescu R, Bejarano M, et al. Sp1 is up-regulated in cellular and transgenic models of Huntington disease, and its reduction is neuroprotective. J Biol Chem. 2006;281:16672–16680. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
