

Protease inhibitors derived from elafin and SLPI and engineered to have enhanced specificity towards neutrophil serine proteases
- PMID: 19241385
- PMCID: PMC2760364
- DOI: 10.1002/pro.64
Protease inhibitors derived from elafin and SLPI and engineered to have enhanced specificity towards neutrophil serine proteases
Abstract
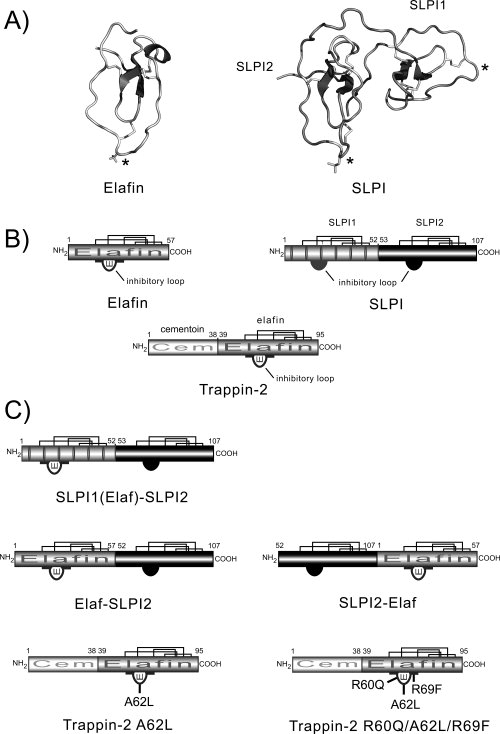
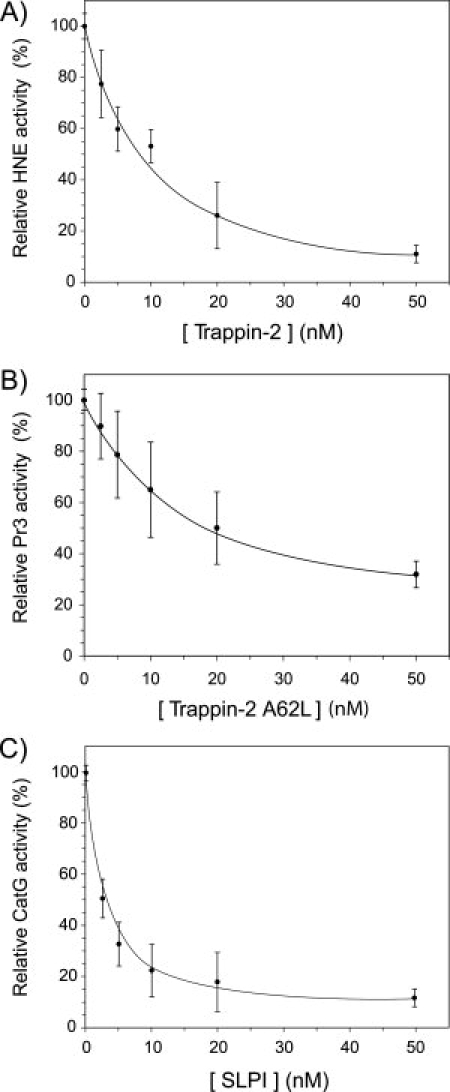
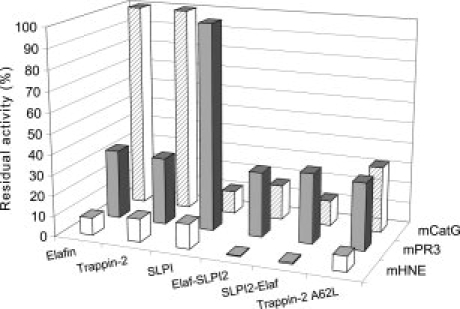
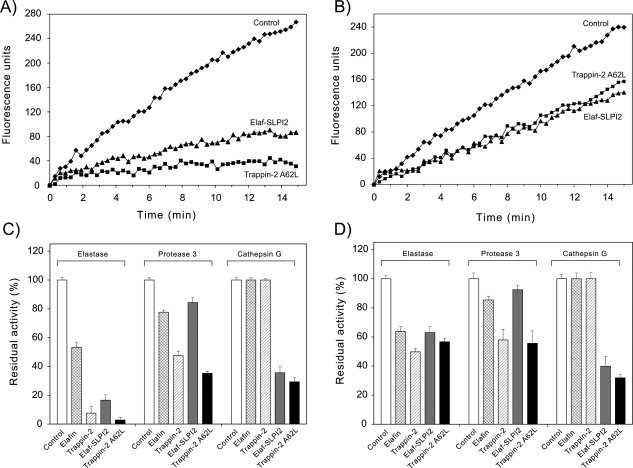
The secretory leukocyte protease inhibitor (SLPI), elafin, and its biologically active precursor trappin-2 are endogeneous low-molecular weight inhibitors of the chelonianin family that control the enzymatic activity of neutrophil serine proteases (NSPs) like elastase, proteinase 3, and cathepsin G. These inhibitors may be of therapeutic value, since unregulated NSP activities are linked to inflammatory lung diseases. However SLPI inhibits elastase and cathepsin G but not proteinase 3, while elafin targets elastase and proteinase 3 but not cathepsin G. We have used two strategies to design polyvalent inhibitors of NSPs that target all three NSPs and may be used in the aerosol-based treatment of inflammatory lung diseases. First, we fused the elafin domain with the second inhibitory domain of SLPI to produce recombinant chimeras that had the inhibitory properties of both parent molecules. Second, we generated the trappin-2 variant, trappin-2 A62L, in which the P1 residue Ala is replaced by Leu, as in the corresponding position in SLPI domain 2. The chimera inhibitors and trappin-2 A62L are tight-binding inhibitors of all three NSPs with subnanomolar K(i)s, similar to those of the parent molecules for their respective target proteases. We have also shown that these molecules inhibit the neutrophil membrane-bound forms of all three NSPs. The trappin-2 A62L and elafin-SLPI chimeras, like wild-type elafin and trappin-2, can be covalently cross-linked to fibronectin or elastin by a tissue transglutaminase, while retaining their polypotent inhibition of NSPs. Therefore, the inhibitors described herein have the appropriate properties to be further evaluated as therapeutic anti-inflammatory agents.
Figures


Similar articles
-
Secretory leukocyte protease inhibitor (SLPI) is, like its homologue trappin-2 (pre-elafin), a transglutaminase substrate.PLoS One. 2011;6(6):e20976. doi: 10.1371/journal.pone.0020976. Epub 2011 Jun 7. PLoS One. 2011. PMID: 21687692 Free PMC article.
-
Protection of lung epithelial cells from protease-mediated injury by trappin-2 A62L, an engineered inhibitor of neutrophil serine proteases.Biochem Pharmacol. 2012 Jun 15;83(12):1663-73. doi: 10.1016/j.bcp.2012.03.009. Epub 2012 Mar 23. Biochem Pharmacol. 2012. PMID: 22465040
-
Multifaceted roles of human elafin and secretory leukocyte proteinase inhibitor (SLPI), two serine protease inhibitors of the chelonianin family.Biochimie. 2008 Feb;90(2):284-95. doi: 10.1016/j.biochi.2007.09.007. Epub 2007 Sep 22. Biochimie. 2008. PMID: 17964057 Review.
-
Kinetics of the inhibition of neutrophil proteinases by recombinant elafin and pre-elafin (trappin-2) expressed in Pichia pastoris.Eur J Biochem. 2004 Jun;271(12):2370-8. doi: 10.1111/j.1432-1033.2004.04156.x. Eur J Biochem. 2004. PMID: 15182352
-
SLPI and trappin-2 as therapeutic agents to target airway serine proteases in inflammatory lung diseases: current and future directions.Biochem Soc Trans. 2011 Oct;39(5):1441-6. doi: 10.1042/BST0391441. Biochem Soc Trans. 2011. PMID: 21936830 Review.
Cited by
-
On the Process of Discovering Leads That Target the Heparin-Binding Site of Neutrophil Elastase in the Sputum of Cystic Fibrosis Patients.J Med Chem. 2019 Jun 13;62(11):5501-5511. doi: 10.1021/acs.jmedchem.9b00379. Epub 2019 May 28. J Med Chem. 2019. PMID: 31074986 Free PMC article.
-
Systemic Markers of Lung Function and Forced Expiratory Volume in 1 Second Decline across Diverse Cohorts.Ann Am Thorac Soc. 2023 Aug;20(8):1124-1135. doi: 10.1513/AnnalsATS.202210-857OC. Ann Am Thorac Soc. 2023. PMID: 37351609 Free PMC article.
-
Secretory leukocyte protease inhibitor (SLPI) is, like its homologue trappin-2 (pre-elafin), a transglutaminase substrate.PLoS One. 2011;6(6):e20976. doi: 10.1371/journal.pone.0020976. Epub 2011 Jun 7. PLoS One. 2011. PMID: 21687692 Free PMC article.
-
Interaction of serine proteases from polymorphonuclear leucocytes with the cell surface and heparin.Inflammation. 2012 Feb;35(1):81-8. doi: 10.1007/s10753-011-9292-x. Inflammation. 2012. PMID: 21246269
-
Genetic correlations, shared risk genes and immunity landscapes between COVID-19 and venous thromboembolism: evidence from GWAS and bulk transcriptome data.Inflamm Res. 2024 Apr;73(4):619-640. doi: 10.1007/s00011-024-01857-w. Epub 2024 Mar 3. Inflamm Res. 2024. PMID: 38433131
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
