

HLA footprints on human immunodeficiency virus type 1 are associated with interclade polymorphisms and intraclade phylogenetic clustering
- PMID: 19244334
- PMCID: PMC2668443
- DOI: 10.1128/JVI.02017-08
HLA footprints on human immunodeficiency virus type 1 are associated with interclade polymorphisms and intraclade phylogenetic clustering
Erratum in
- J Virol. 2011 May;85(9):4635
Abstract
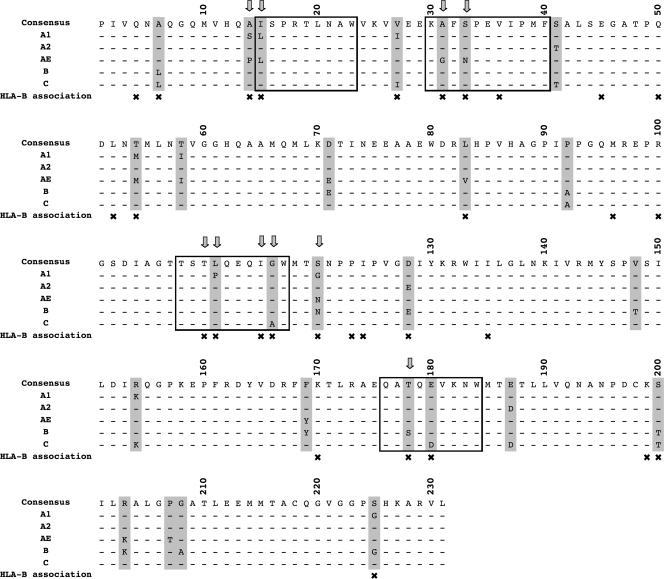
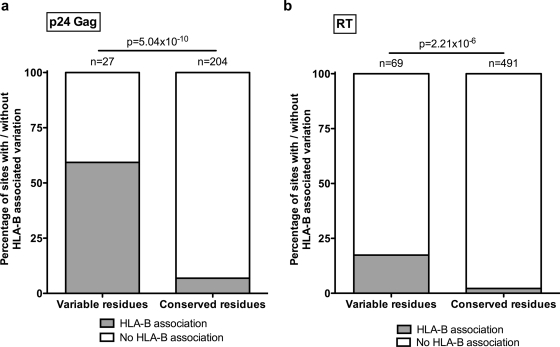
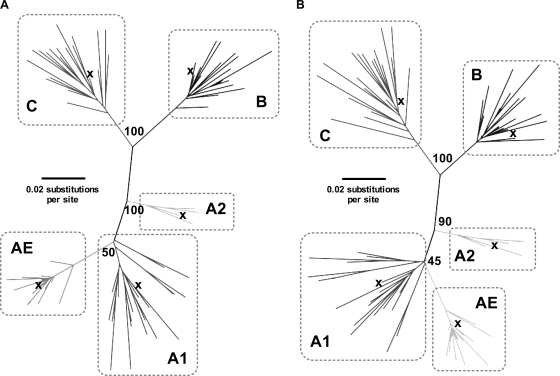
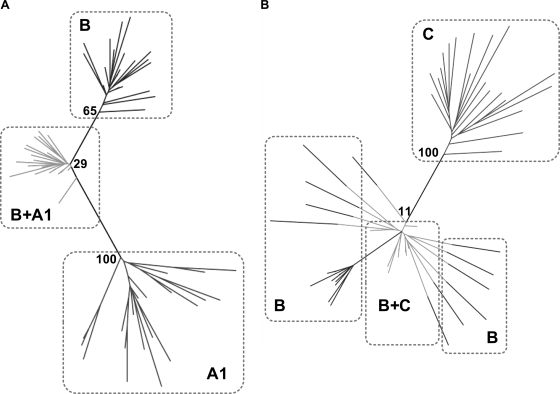
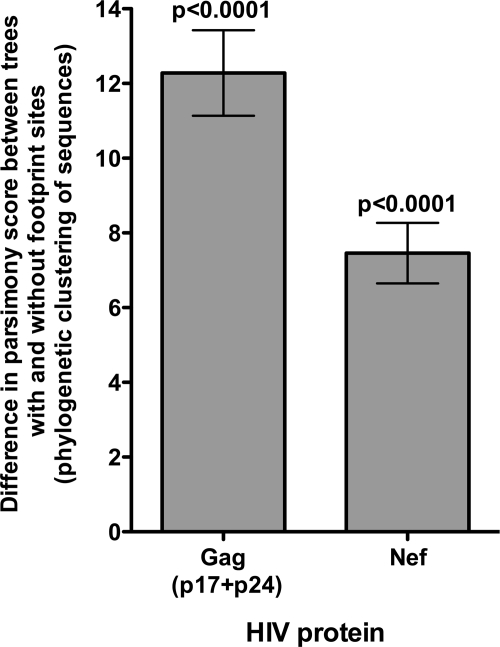
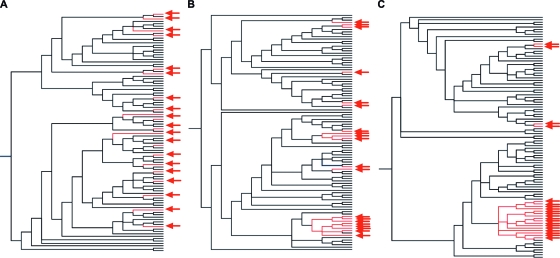
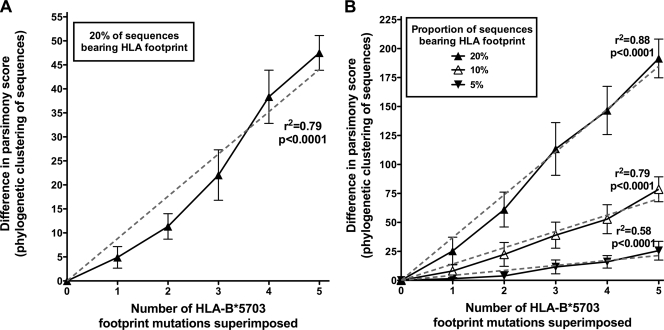
The selection of escape mutations has a major impact on immune control of infections with viruses such as human immunodeficiency virus (HIV). Viral evasion of CD8(+) T-cell responses leaves predictable combinations of escape mutations, termed HLA "footprints." The most clearly defined footprints are those associated with HLA alleles that are linked with successful control of HIV, such as HLA-B*57. Here we investigated the extent to which HLA footprint sites in HIV type 1 (HIV-1) are associated with viral evolution among and within clades. First, we examined the extent to which amino acid differences between HIV-1 clades share identity with sites of HLA-mediated selection pressure and observed a strong association, in particular with respect to sites of HLA-B selection (P < 10(-6)). Similarly, the sites of amino acid variability within a clade were found to overlap with sites of HLA-selected mutation. Second, we studied the impact of HLA selection on interclade phylogeny. Removing the sites of amino acid variability did not significantly affect clade-specific clustering, reflecting the central role of founder effects in establishing distinct clades. However, HLA footprints may underpin founder strains, and we show that amino acid substitutions between clades alter phylogeny, underlining a potentially substantial role for HLA in driving ongoing viral evolution. Finally, we investigated the impact of HLA selection on within-clade phylogeny and demonstrate that even a single HLA allele footprint can result in significant phylogenetic clustering of sequences. In conclusion, these data highlight the fact that HLA can be a strong selection force for both intra- and interclade HIV evolution at a population level.
Figures
Similar articles
-
Weaker HLA Footprints on HIV in the Unique and Highly Genetically Admixed Host Population of Mexico.J Virol. 2018 Jan 2;92(2):e01128-17. doi: 10.1128/JVI.01128-17. Print 2018 Jan 15. J Virol. 2018. PMID: 29093100 Free PMC article.
-
Central role of reverting mutations in HLA associations with human immunodeficiency virus set point.J Virol. 2008 Sep;82(17):8548-59. doi: 10.1128/JVI.00580-08. Epub 2008 Jul 2. J Virol. 2008. PMID: 18596105 Free PMC article.
-
Transmission and accumulation of CTL escape variants drive negative associations between HIV polymorphisms and HLA.J Exp Med. 2005 Mar 21;201(6):891-902. doi: 10.1084/jem.20041455. J Exp Med. 2005. PMID: 15781581 Free PMC article.
-
Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1.PLoS Pathog. 2007 Jul;3(7):e94. doi: 10.1371/journal.ppat.0030094. PLoS Pathog. 2007. PMID: 17616974 Free PMC article.
-
Identification of HLA class I-associated amino acid polymorphisms in the HIV-1C proteome.AIDS Res Hum Retroviruses. 2007 Jan;23(1):165-74. doi: 10.1089/aid.2006.0131. AIDS Res Hum Retroviruses. 2007. PMID: 17263647
Cited by
-
CD8+ T Cell Breadth and Ex Vivo Virus Inhibition Capacity Distinguish between Viremic Controllers with and without Protective HLA Class I Alleles.J Virol. 2016 Jul 11;90(15):6818-6831. doi: 10.1128/JVI.00276-16. Print 2016 Aug 1. J Virol. 2016. PMID: 27194762 Free PMC article.
-
Adaptive evolution of HIV at HLA epitopes is associated with ethnicity in Canada.PLoS One. 2012;7(5):e36933. doi: 10.1371/journal.pone.0036933. Epub 2012 May 31. PLoS One. 2012. PMID: 22693560 Free PMC article.
-
The antiviral efficacy of HIV-specific CD8⁺ T-cells to a conserved epitope is heavily dependent on the infecting HIV-1 isolate.PLoS Pathog. 2011 May;7(5):e1001341. doi: 10.1371/journal.ppat.1001341. Epub 2011 May 12. PLoS Pathog. 2011. PMID: 21589893 Free PMC article.
-
Selection pressure in CD8⁺ T-cell epitopes in the pol gene of HIV-1 infected individuals in Colombia. A bioinformatic approach.Viruses. 2015 Mar 20;7(3):1313-31. doi: 10.3390/v7031313. Viruses. 2015. PMID: 25803098 Free PMC article.
-
Pressure from TRIM5α contributes to control of HIV-1 replication by individuals expressing protective HLA-B alleles.J Virol. 2013 Sep;87(18):10368-80. doi: 10.1128/JVI.01313-13. Epub 2013 Jul 17. J Virol. 2013. PMID: 23864638 Free PMC article.
References
-
- Allen, T. M., M. Altfeld, S. C. Geer, E. T. Kalife, C. Moore, M. O'Sullivan, K., I. Desouza, M. E. Feeney, R. L. Eldridge, E. L. Maier, D. E. Kaufmann, M. P. Lahaie, L. Reyor, G. Tanzi, M. N. Johnston, C. Brander, R. Draenert, J. K. Rockstroh, H. Jessen, E. S. Rosenberg, S. A. Mallal, and B. D. Walker. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J. Virol. 7913239-13249. - PMC - PubMed
-
- Betts, M. R., B. Exley, D. A. Price, A. Bansal, Z. T. Camacho, V. Teaberry, S. M. West, D. R. Ambrozak, G. Tomaras, M. Roederer, J. M. Kilby, J. Tartaglia, R. Belshe, F. Gao, D. C. Douek, K. J. Weinhold, R. A. Koup, P. Goepfert, and G. Ferrari. 2005. Characterization of functional and phenotypic changes in anti-Gag vaccine-induced T cell responses and their role in protection after HIV-1 infection. Proc. Natl. Acad. Sci. USA 1024512-4517. - PMC - PubMed
-
- Bhattacharya, T., M. Daniels, D. Heckerman, B. Foley, N. Frahm, C. Kadie, J. Carlson, K. Yusim, B. McMahon, B. Gaschen, S. Mallal, J. I. Mullins, D. C. Nickle, J. Herbeck, C. Rousseau, G. H. Learn, T. Miura, C. Brander, B. Walker, and B. Korber. 2007. Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science 3151583-1586. - PubMed
-
- Brockman, M. A., A. Schneidewind, M. Lahaie, A. Schmidt, T. Miura, I. Desouza, F. Ryvkin, C. A. Derdeyn, S. Allen, E. Hunter, J. Mulenga, P. A. Goepfert, B. D. Walker, and T. M. Allen. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J. Virol. 8112608-12618. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
