

Targeting the Notch1 and mTOR pathways in a mouse T-ALL model
- PMID: 19246562
- PMCID: PMC2699237
- DOI: 10.1182/blood-2008-02-136762
Targeting the Notch1 and mTOR pathways in a mouse T-ALL model
Abstract
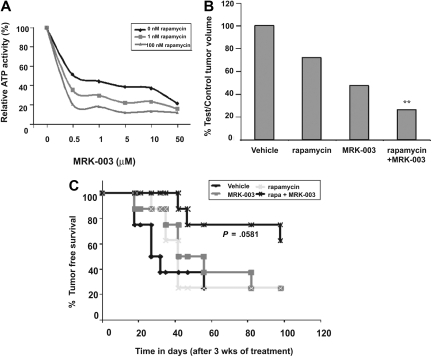
Mutations in NOTCH1 are frequently detected in patients with T-cell acute lymphoblastic leukemia (T-ALL) and in mouse T-ALL models. Treatment of mouse or human T-ALL cell lines in vitro with gamma-secretase inhibitors (GSIs) results in growth arrest and/or apoptosis. These studies suggest GSIs as potential therapeutic agents in the treatment of T-ALL. To determine whether GSIs have antileukemic activity in vivo, we treated near-end-stage Tal1/Ink4a/Arf+/- leukemic mice with vehicle or with a GSI developed by Merck (MRK-003). We found that GSI treatment significantly extended the survival of leukemic mice compared with vehicle-treated mice. Notch1 target gene expression was repressed and increased numbers of apoptotic cells were observed in the GSI-treated mice, demonstrating that Notch1 inhibition in vivo induces apoptosis. T-ALL cell lines also exhibit PI3K/mTOR pathway activation, indicating that rapamycin may also have therapeutic benefit. When GSIs are administered in combination with rapamycin, mTOR kinase activity is ablated and apoptosis induced. Moreover, GSI and rapamycin treatment inhibits human T-ALL growth and extends survival in a mouse xenograft model. This work supports the idea of targeting NOTCH1 in T-ALL and suggests that inhibition of the mTOR and NOTCH1 pathways may have added efficacy.
Figures






Comment in
-
Notch targeting 2.0.Blood. 2009 Jun 11;113(24):6044-5. doi: 10.1182/blood-2009-03-212829. Blood. 2009. PMID: 19520812 No abstract available.
References
-
- Aplan PD, Lombardi DP, Reaman GH, Sather HN, Hammond GD, Kirsch IR. Involvement of the putative hematopoietic transcription factor SCL in T-cell acute lymphoblastic leukemia. Blood. 1992;79:1327–1333. - PubMed
-
- Bash RO, Hall S, Timmons CF, et al. Does activation of the TAL1 gene occur in a majority of patients with T-cell acute lymphoblastic leukemia? a pediatric oncology group study. Blood. 1995;86:666–676. - PubMed
-
- Bernard O, Lecointe N, Jonveaux P, et al. Two site-specific deletions and t(1;14) translocation restricted to human T-cell acute leukemias disrupt the 5′ part of the tal-1 gene. Oncogene. 1991;6:1477–1488. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
