

Chordin is a modifier of tbx1 for the craniofacial malformations of 22q11 deletion syndrome phenotypes in mouse
- PMID: 19247433
- PMCID: PMC2640462
- DOI: 10.1371/journal.pgen.1000395
Chordin is a modifier of tbx1 for the craniofacial malformations of 22q11 deletion syndrome phenotypes in mouse
Abstract
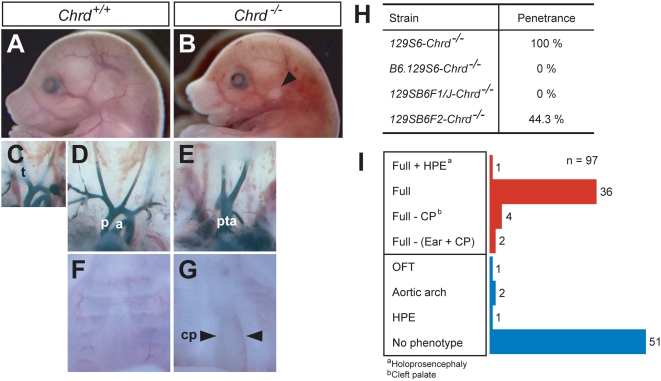
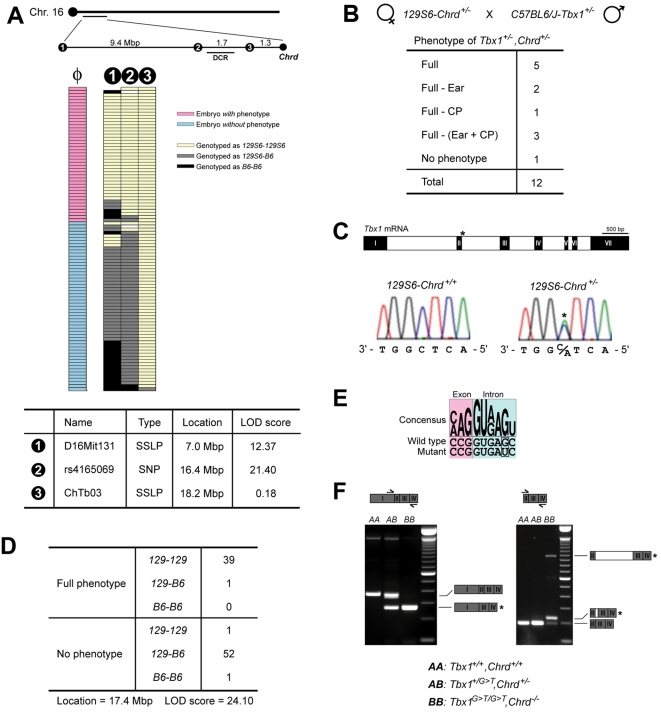
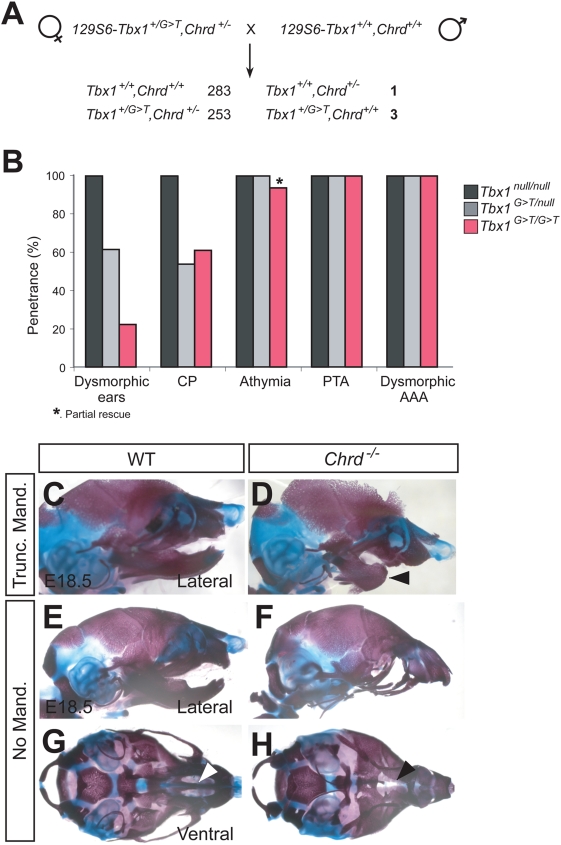
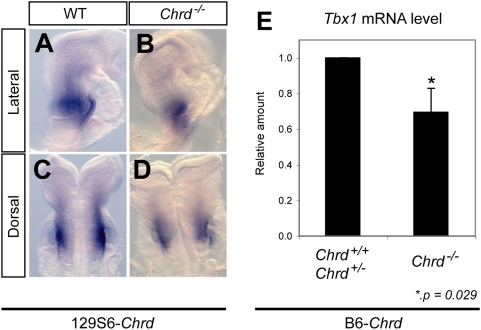
Point mutations in TBX1 can recapitulate many of the structural defects of 22q11 deletion syndromes (22q11DS), usually associated with a chromosomal deletion at 22q1.2. 22q11DS often includes specific cardiac and pharyngeal organ anomalies, but the presence of characteristic craniofacial defects is highly variable. Even among family members with a single TBX1 point mutation but no cytological deletion, cleft palate and low-set ears may or may not be present. In theory, such differences could depend on an unidentified, second-site lesion that modifies the craniofacial consequences of TBX1 deficiency. We present evidence for such a locus in a mouse model. Null mutations of chordin have been reported to cause severe defects recapitulating 22q11DS, which we show are highly dependent on genetic background. In an inbred strain in which chordin(-/-) is fully penetrant, we found a closely linked, strong modifier--a mutation in a Tbx1 intron causing severe splicing defects. Without it, lack of chordin results in a low penetrance of mandibular hypoplasia but no cardiac or thoracic organ malformations. This hypomorphic Tbx1 allele per se results in defects resembling 22q11DS but with a low penetrance of hallmark craniofacial malformations, unless chordin is mutant. Thus, chordin is a modifier for the craniofacial anomalies of Tbx1 mutations, demonstrating the existence of a second-site modifier for a specific subset of the phenotypes associated with 22q11DS.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Paylor R, Lindsay E. Mouse models of 22q11 deletion syndrome. Biol Psychiatry. 2006;59:1172–1179. - PubMed
-
- Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. - PubMed
-
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. - PubMed
-
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
