

CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup
- PMID: 19249009
- PMCID: PMC2668007
- DOI: 10.1016/j.ajhg.2009.02.003
CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup
Abstract
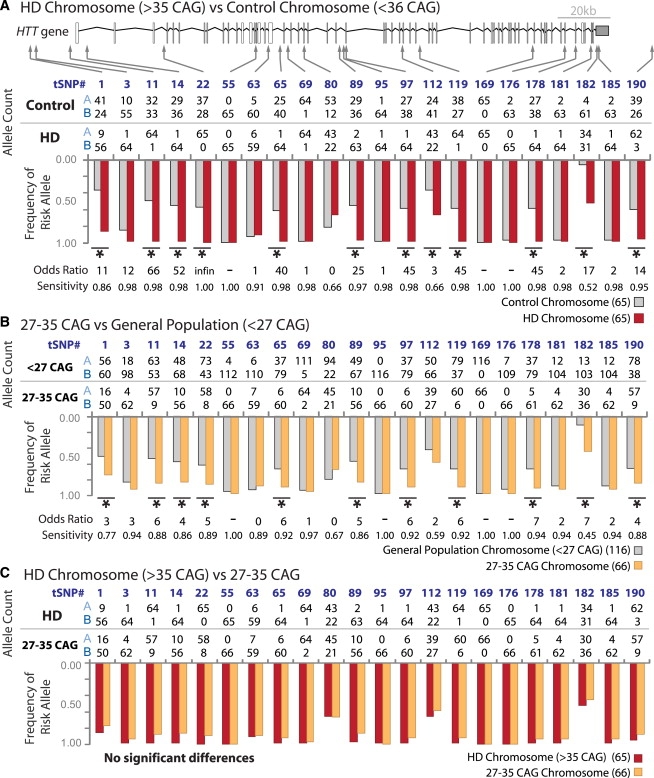
Huntington disease (HD) is an autosomal-dominant disorder that results from >or=36 CAG repeats in the HD gene (HTT). Approximately 10% of patients inherit a chromosome that underwent CAG expansion from an unaffected parent with <36 CAG repeats. This study is a comprehensive analysis of genetic diversity in HTT and reveals that HD patients of European origin (n = 65) have a significant enrichment (95%) of a specific set of 22 tagging single nucleotide polymorphisms (SNPs) that constitute a single haplogroup. The disease association of many SNPs is much stronger than any previously reported polymorphism and was confirmed in a replication cohort (n = 203). Importantly, the same haplogroup is also significantly enriched (83%) in individuals with 27-35 CAG repeats (intermediate alleles, n = 66), who are unaffected by the disease, but have increased CAG tract sizes relative to the general population (n = 116). These data support a stepwise model for CAG expansion into the affected range (>or=36 CAG) and identifies specific haplogroup variants in the general population associated with this instability. The specific variants at risk for CAG expansion are not present in the general population in China, Japan, and Nigeria where the prevalence of HD is much lower. The current data argue that cis-elements have a major predisposing influence on CAG instability in HTT. The strong association between specific SNP alleles and CAG expansion also provides an opportunity of personalized therapeutics in HD where the clinical development of only a small number of allele-specific targets may be sufficient to treat up to 88% of the HD patient population.
Figures






Comment in
-
Haplotype background, repeat length evolution, and Huntington's disease.Am J Hum Genet. 2009 Dec;85(6):939-42. doi: 10.1016/j.ajhg.2009.11.002. Am J Hum Genet. 2009. PMID: 20004772 Free PMC article. No abstract available.
References
-
- Kremer B., Goldberg P., Andrew S.E., Theilmann J., Telenius H., Zeisler J., Squitieri F., Lin B., Bassett A., Almqvist E. A worldwide study of the Huntington's disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 1994;330:1401–1406. - PubMed
-
- Walker F.O. Huntington's disease. Lancet. 2007;369:218–228. - PubMed
-
- Telenius H., Kremer H.P., Theilmann J., Andrew S.E., Almqvist E., Anvret M., Greenberg C., Greenberg J., Lucotte G., Squitieri F. Molecular analysis of juvenile Huntington disease: the major influence on (CAG)n repeat length is the sex of the affected parent. Hum. Mol. Genet. 1993;2:1535–1540. - PubMed
-
- Almqvist E.W., Elterman D.S., MacLeod P.M., Hayden M.R. High incidence rate and absent family histories in one quarter of patients newly diagnosed with Huntington disease in British Columbia. Clin. Genet. 2001;60:198–205. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
