

Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice
- PMID: 19251638
- PMCID: PMC2657393
- DOI: 10.1073/pnas.0813339106
Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice
Abstract
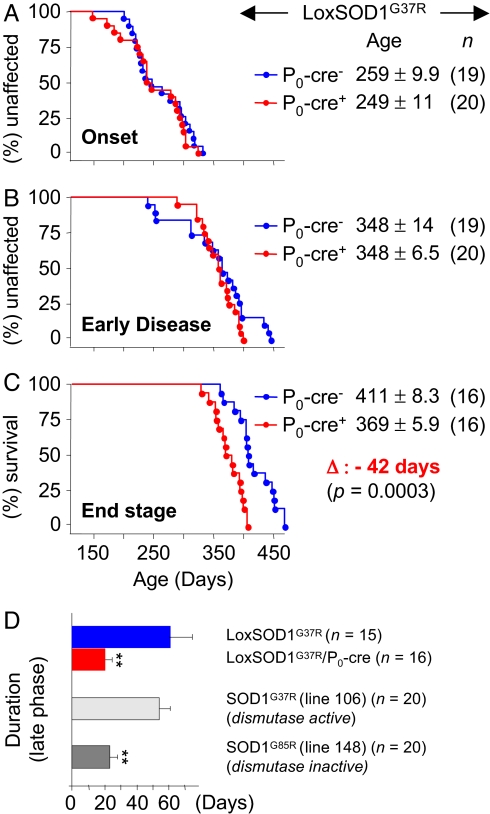
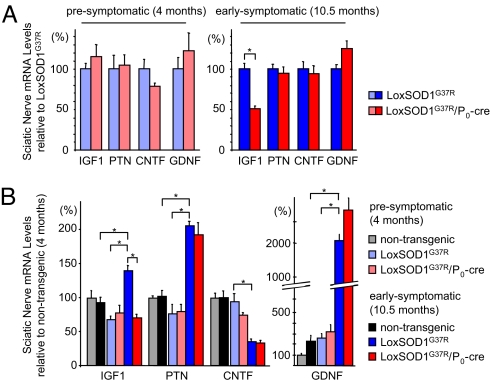
Neurodegeneration in an inherited form of ALS is non-cell-autonomous, with ALS-causing mutant SOD1 damage developed within multiple cell types. Selective inactivation within motor neurons of an ubiquitously expressed mutant SOD1 gene has demonstrated that mutant damage within motor neurons is a determinant of disease initiation, whereas mutant synthesis within neighboring astrocytes or microglia accelerates disease progression. We now report the surprising finding that diminished synthesis (by 70%) within Schwann cells of a fully dismutase active ALS-linked mutant (SOD1(G37R)) significantly accelerates disease progression, accompanied by reduction of insulin-like growth factor 1 (IGF-1) in nerves. Coupled with shorter disease duration in mouse models caused by dismutase inactive versus dismutase active SOD1 mutants, our findings implicate an oxidative cascade during disease progression that is triggered within axon ensheathing Schwann cells and that can be ameliorated by elevated dismutase activity. Thus, therapeutic down-regulation of dismutase active mutant SOD1 in familial forms of ALS should be targeted away from Schwann cells.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Boillee S, Vande Velde C, Cleveland DW. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. - PubMed
-
- McNamara JO, Fridovich I. Human genetics. Did radicals strike Lou Gehrig? Nature. 1993;362:20–21. - PubMed
-
- Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. - PubMed
-
- Liu J, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. - PubMed
-
- Urushitani M, et al. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–118. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
