

Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation
- PMID: 19251646
- PMCID: PMC2649209
- DOI: 10.1073/pnas.0810053106
Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation
Abstract
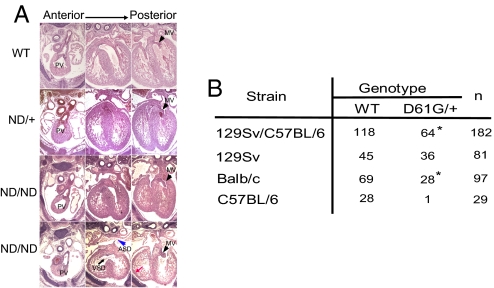
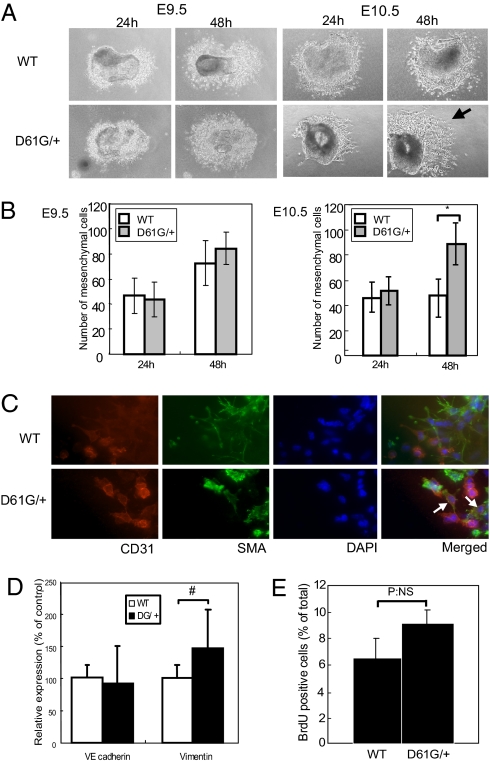
Noonan syndrome (NS), the most common single-gene cause of congenital heart disease, is an autosomal dominant disorder that also features proportionate short stature, facial abnormalities, and an increased risk of myeloproliferative disease. Germline-activating mutations in PTPN11, which encodes the protein tyrosine phosphatase SHP2, cause about half of NS cases; other causative alleles include KRAS, SOS1, and RAF1 mutants. We showed previously that knock-in mice bearing the NS mutant Ptpn11(D61G) on a mixed 129S4/SvJae X C57BL6/J background exhibit all major NS features, including a variety of cardiac defects, with variable penetrance. However, the cellular and molecular mechanisms underlying NS cardiac defects and whether genetic background and/or the specific NS mutation contribute to the NS phenotype remained unclear. Here, using an inducible knock-in approach, we show that all cardiac defects in NS result from mutant Shp2 expression in the endocardium, not in the myocardium or neural crest. Furthermore, the penetrance of NS defects is affected by genetic background and the specific Ptpn11 allele. Finally, ex vivo assays and pharmacological approaches show that NS mutants cause cardiac valve defects by increasing Erk MAPK activation, probably downstream of ErbB family receptor tyrosine kinases, extending the interval during which cardiac endocardial cells undergo endocardial-mesenchymal transformation. Our data provide a mechanistic underpinning for the cardiac defects in this disorder.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Noonan JA. Noonan syndrome. An update and review for the primary pediatrician. Clinical Pediatrics. 1994;33:548–555. - PubMed
-
- Tartaglia M, Gelb BD. Noonan syndrome and related disorders: Genetics and pathogenesis. Annual Review of Genomics and Human Genetics. 2005;6:45–68. - PubMed
-
- Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;27:179–192. - PubMed
-
- Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–293. - PubMed
-
- Bentires-Alj M, Kontaridis MI, Neel BG. Stops along the RAS pathway in human genetic disease. Nat Med. 2006;12:283–285. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
