

(V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway
- PMID: 19251651
- PMCID: PMC2649208
- DOI: 10.1073/pnas.0900780106
(V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway
Abstract
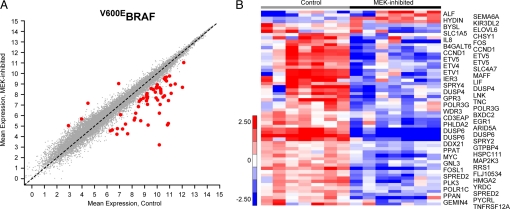
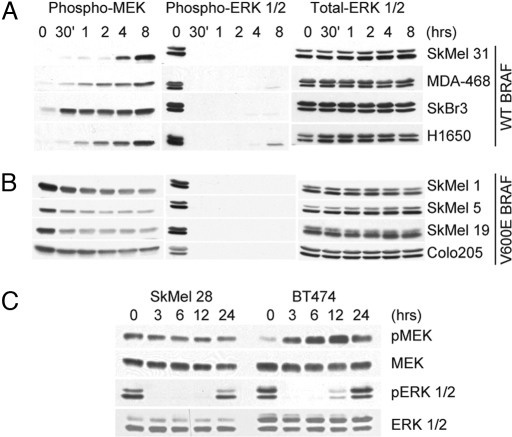
Tumors with mutant BRAF and those with receptor tyrosine kinase (RTK) activation have similar levels of phosphorylated ERK, but only the former depend on ERK signaling for proliferation. The mitogen-activated protein kinase, extracellular signal-regulated kinase kinase (MEK)/ERK-dependent transcriptional output was defined as the genes whose expression changes significantly 8 h after MEK inhibition. In (V600E)BRAF cells, this output is comprised of 52 genes, including transcription factors that regulate transformation and members of the dual specificity phosphatase and Sprouty gene families, feedback inhibitors of ERK signaling. No such genes were identified in RTK tumor cells, suggesting that ERK pathway signaling output is selectively activated in BRAF mutant tumors. We find that RAF signaling is feedback down-regulated in RTK cells, but is insensitive to this feedback in BRAF mutant tumors. Physiologic feedback inhibition of RAF/MEK signaling down-regulates ERK output in RTK cells; evasion of this feedback in mutant BRAF cells is associated with increased transcriptional output and MEK/ERK-dependent transformation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
-
- Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. - PubMed
-
- Hoshino R, et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene. 1999;18:813–822. - PubMed
-
- Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. - PubMed
-
- Brose MS, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. - PubMed
-
- Gorden A, et al. Analysis of BRAF and N-RAS mutations in metastatic melanoma tissues. Cancer Res. 2003;63:3955–3957. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
