

Longitudinal changes of mtDNA A3243G mutation load and level of functioning in MELAS
- PMID: 19253345
- PMCID: PMC2663596
- DOI: 10.1002/ajmg.a.32703
Longitudinal changes of mtDNA A3243G mutation load and level of functioning in MELAS
Abstract
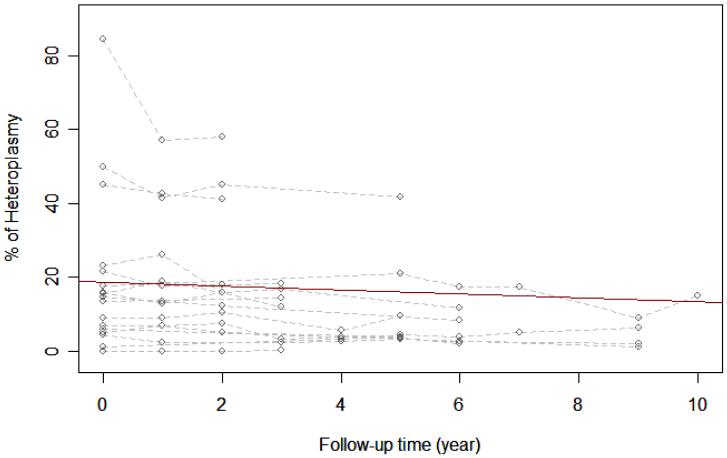
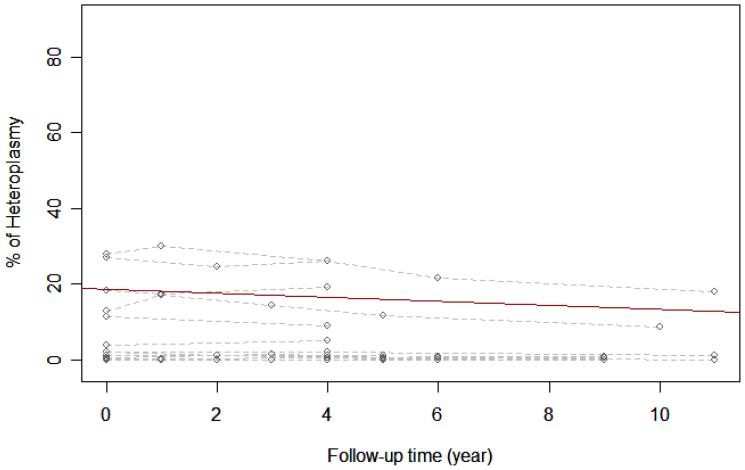
Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), one of the most common mitochondrial multisystemic diseases, is most commonly associated with an A-to-G transition at nucleotide position 3243 (A3243G) in mitochondrial DNA. We studied 34 individuals harboring the A3243G mutation for up to 7 years; 17 had the full MELAS phenotype and 17 who were classified as "carrier relatives" because they were either asymptomatic or had some symptoms suggestive of mitochondrial disease but no seizures or strokes. Using the sensitive real-time polymerase chain reaction to quantify the A3243G mutation, we confirmed that the percent mutation decreases progressively in DNA isolated from blood: the average percent decrease was 0.5% per year for fully symptomatic patients and 0.2% per year for oligosymptomatic carrier relatives. We also correlated mutant loads with functional status estimated by the Karnofksky score: even though the mutation load decreases, the level of functioning worsens in fully symptomatic patients, whereas the level of functioning of carrier relatives remains largely unchanged. This study suggests that A3243G mutant load in DNA isolated from blood is neither useful for prognosis nor for functional assessment.
Figures
References
-
- Campos Y, Bautista J, Gutierrez-Rivas E, Chinchon D, Cabello A, Segura D, Arenas J. Clinical heterogeneity in two pedigrees with the 3243 bp tRNA(leu(UUR)) mutation of mitochondrial DNA. Acta Neurol Scand. 1995;91:62–65. - PubMed
-
- Chinnery P, Howell N, Lightowlers R, Turnbull D. The relationship between mutation load and clinical phenotypes. Brain. 1997;120:1713–1721. - PubMed
-
- Ciafaloni E, Ricci E, Servidei S, Shanske S, Silvestri G, Manfredi G, Schon EA, DiMauro S. Widespread tissue distribution of a tRNALeu(UUR) mutation in the mitochondrial DNA of a patient with MELAS syndrome. Neurology. 1991;41:1663–1664. - PubMed
-
- DiMauro S, Moraes CT. Mitochondrial encephalomyopathies. Arch Neurol. 1993;50:1197–1208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
